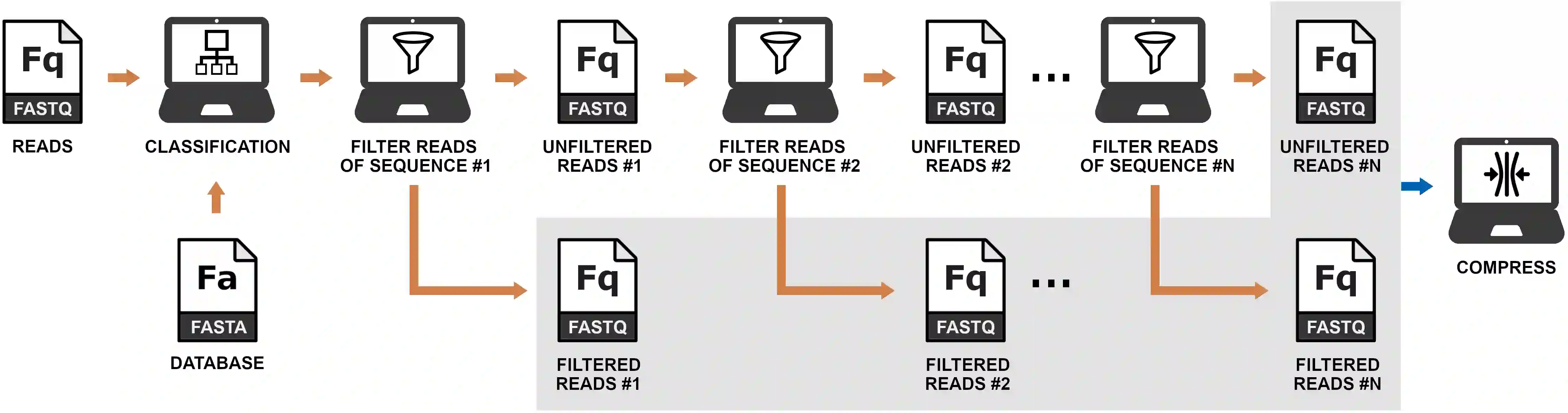
Minimizing data storage poses a significant challenge in large-scale metagenomic projects. In this paper, we present a new method for improving the encoding of FASTQ files generated by metagenomic sequencing. This method incorporates metagenomic classification followed by a recursive filter for clustering reads by DNA sequence similarity to improve the overall reference-free compression. In the results, we show an overall improvement in the compression of several datasets. As hypothesized, we show a progressive compression gain for higher coverage depth and number of identified species. Additionally, we provide an implementation that is freely available at https://github.com/cobilab/mizar and can be customized to work with other FASTQ compression tools.
翻译:数据存储最小化是大型宏基因组项目面临的一项重大挑战。本文提出了一种新方法,用于改善宏基因组测序产生的FASTQ文件的编码。该方法结合宏基因组分类,随后通过递归滤波器按DNA序列相似性对读段进行聚类,以提升整体无参考压缩性能。结果显示,该方法在多个数据集上实现了压缩效果的全面改善。正如我们所假设的,随着测序覆盖深度和识别物种数量的增加,压缩增益呈现渐进式提升。此外,我们提供了可自由获取的实现代码(https://github.com/cobilab/mizar),并支持与其他FASTQ压缩工具进行定制化协同使用。