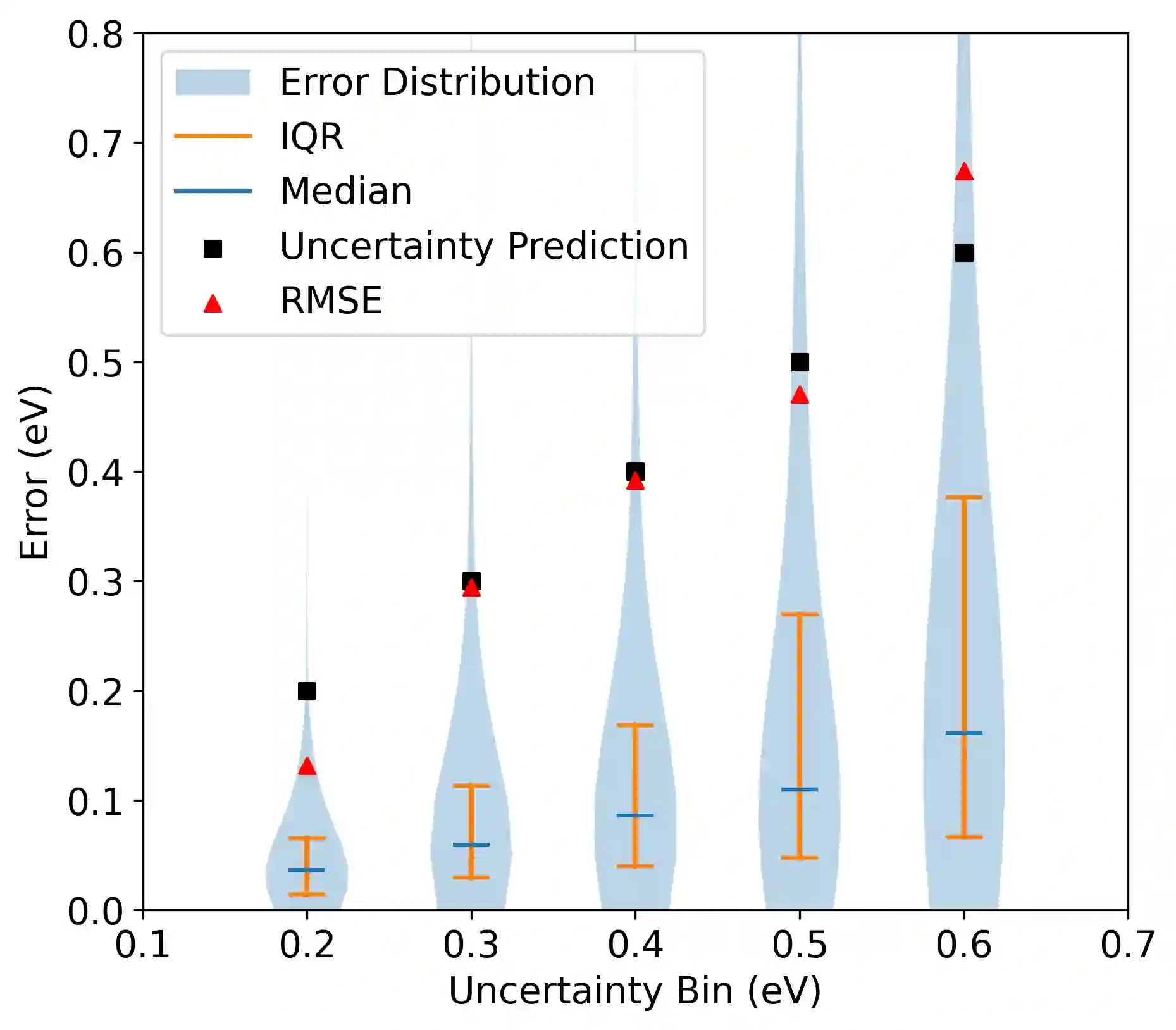
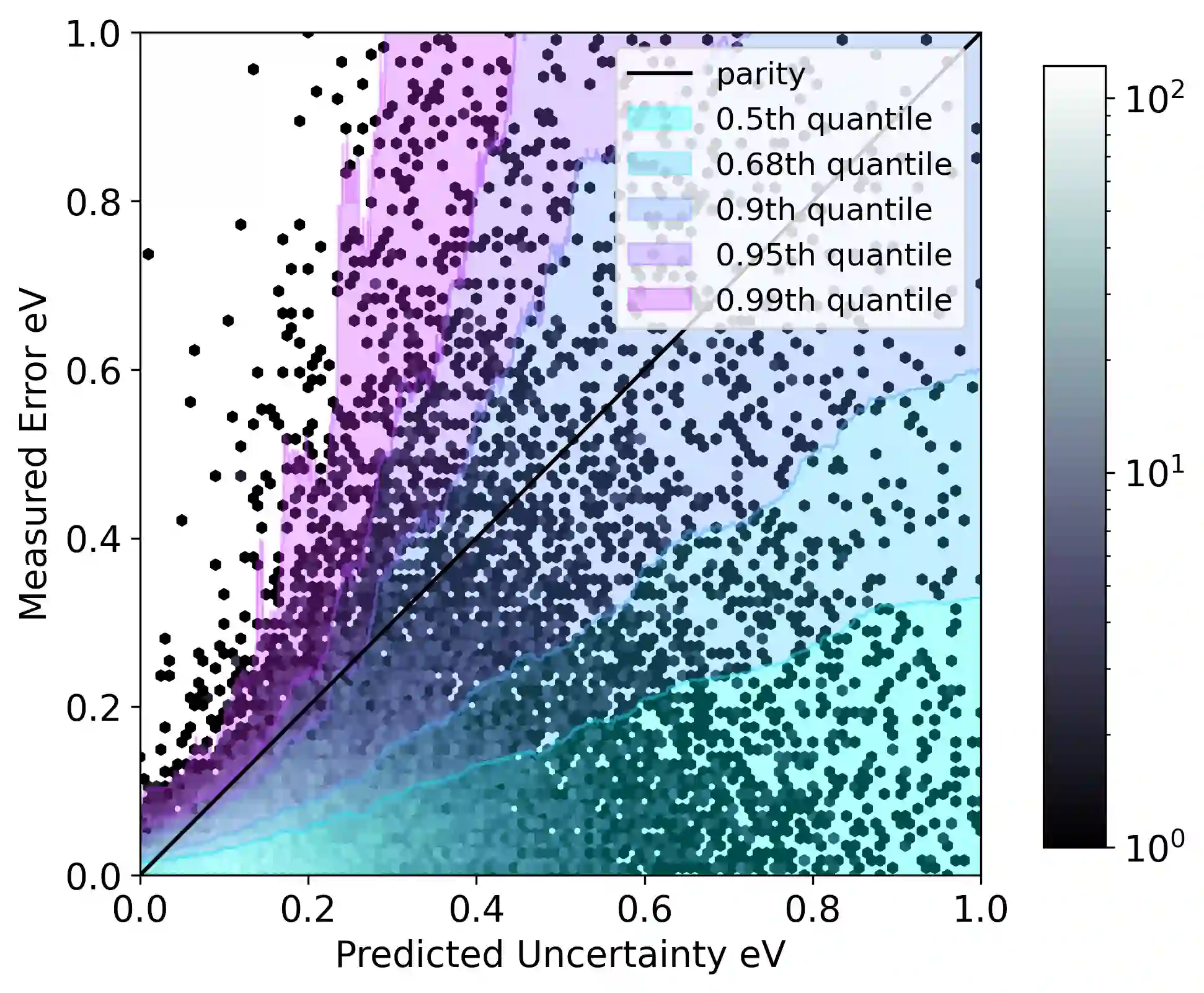
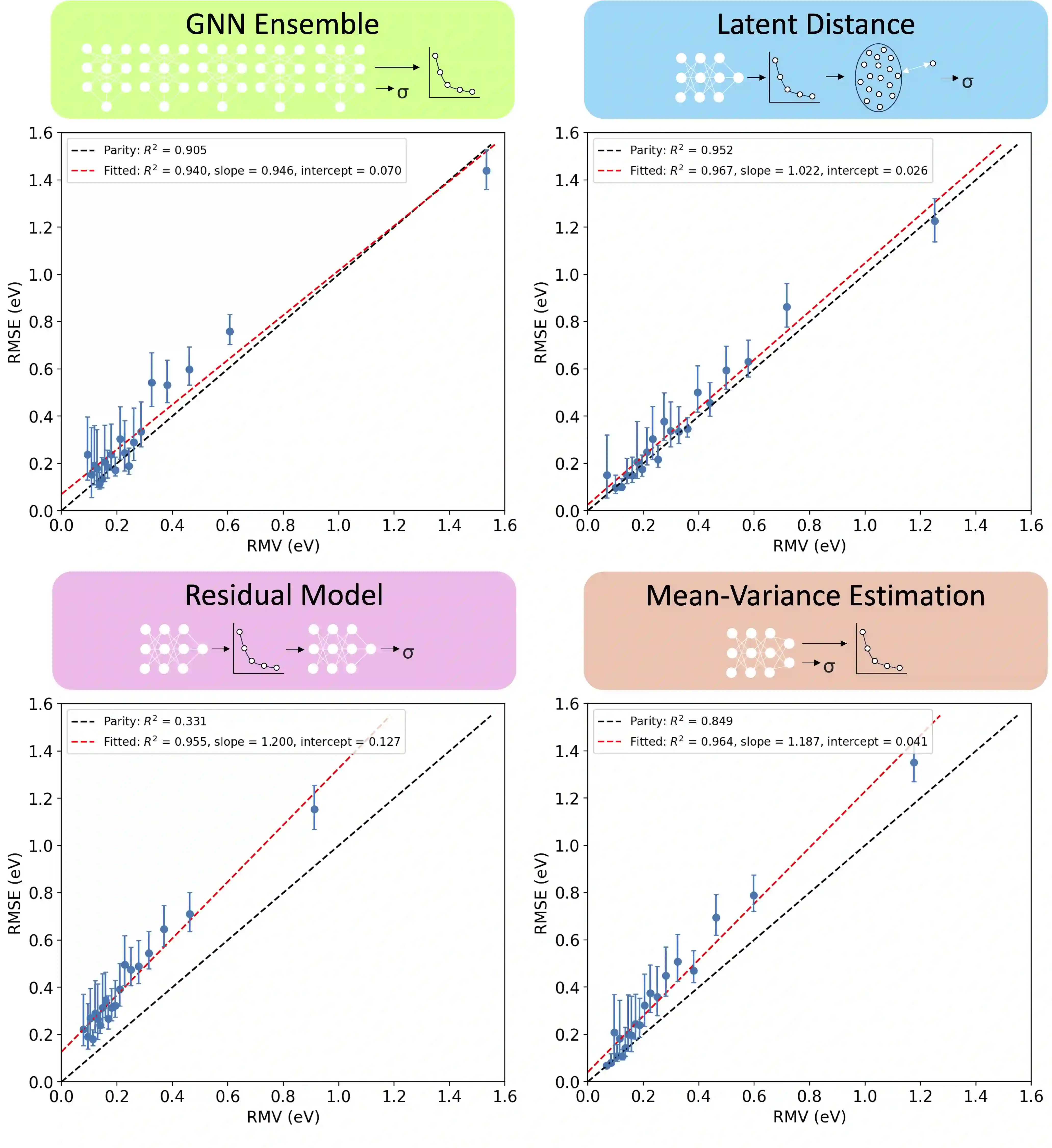
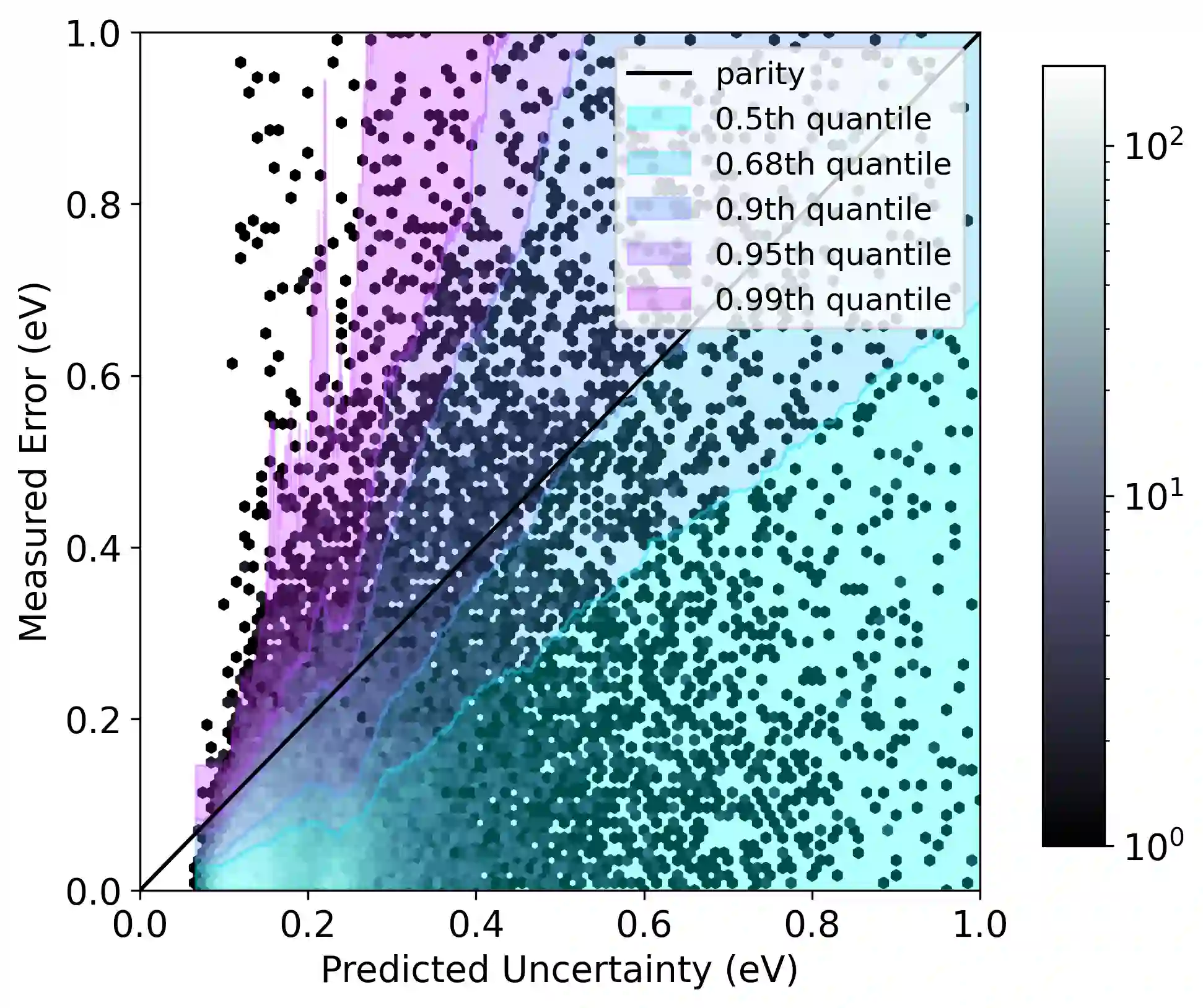
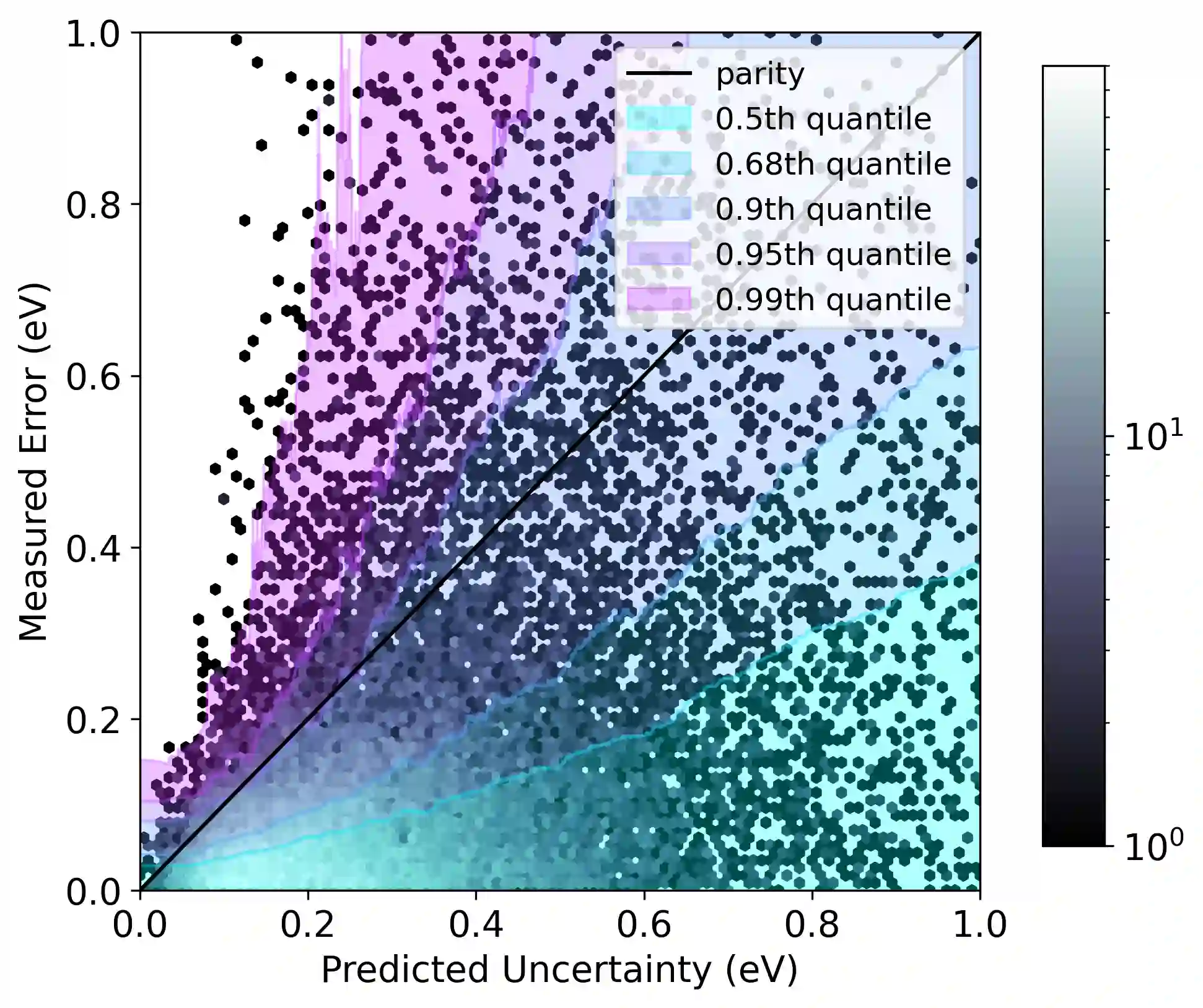
Graph neural networks (GNNs) have been shown to be astonishingly capable models for molecular property prediction, particularly as surrogates for expensive density functional theory calculations of relaxed energy for novel material discovery. However, one limitation of GNNs in this context is the lack of useful uncertainty prediction methods, as this is critical to the material discovery pipeline. In this work, we show that uncertainty quantification for relaxed energy calculations is more complex than uncertainty quantification for other kinds of molecular property prediction, due to the effect that structure optimizations have on the error distribution. We propose that distribution-free techniques are more useful tools for assessing calibration, recalibrating, and developing uncertainty prediction methods for GNNs performing relaxed energy calculations. We also develop a relaxed energy task for evaluating uncertainty methods for equivariant GNNs, based on distribution-free recalibration and using the Open Catalyst Project dataset. We benchmark a set of popular uncertainty prediction methods on this task, and show that latent distance methods, with our novel improvements, are the most well-calibrated and economical approach for relaxed energy calculations. Finally, we demonstrate that our latent space distance method produces results which align with our expectations on a clustering example, and on specific equation of state and adsorbate coverage examples from outside the training dataset.
翻译:图神经网络(GNNs)已被证明是分子性质预测中能力惊人的模型,尤其可作为昂贵密度泛函理论计算在新材料发现中弛豫能量的替代模型。然而,在此背景下GNNs的一个局限是缺乏有效的不确定性预测方法,而这对于材料发现流程至关重要。本研究表明,由于结构优化对误差分布的影响,弛豫能量计算的不确定性量化比其他类型的分子性质预测的不确定性量化更为复杂。我们提出,无分布技术是评估校准度、重新校准以及为执行弛豫能量计算的GNNs开发不确定性预测方法的更有用工具。基于无分布重新校准并利用开放催化剂项目数据集,我们还开发了一个用于评估等变GNNs不确定性方法的弛豫能量任务。我们在此任务上对一系列流行的不确定性预测方法进行基准测试,结果表明采用我们新颖改进的潜在距离方法是弛豫能量计算中校准度最佳且最经济的方法。最后,我们通过聚类示例以及训练数据集外部的特定状态方程和吸附质覆盖度示例,证明我们的潜在空间距离方法产生的结果符合理论预期。