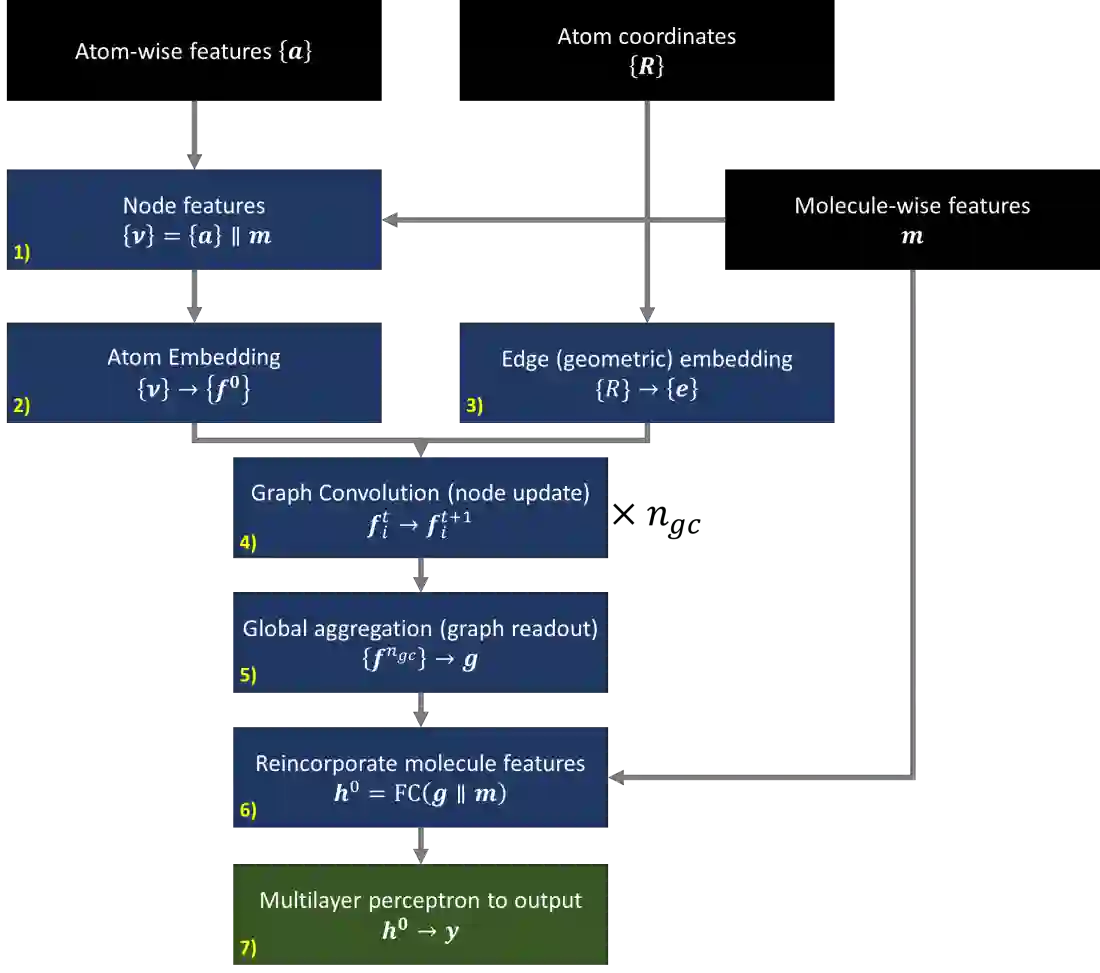
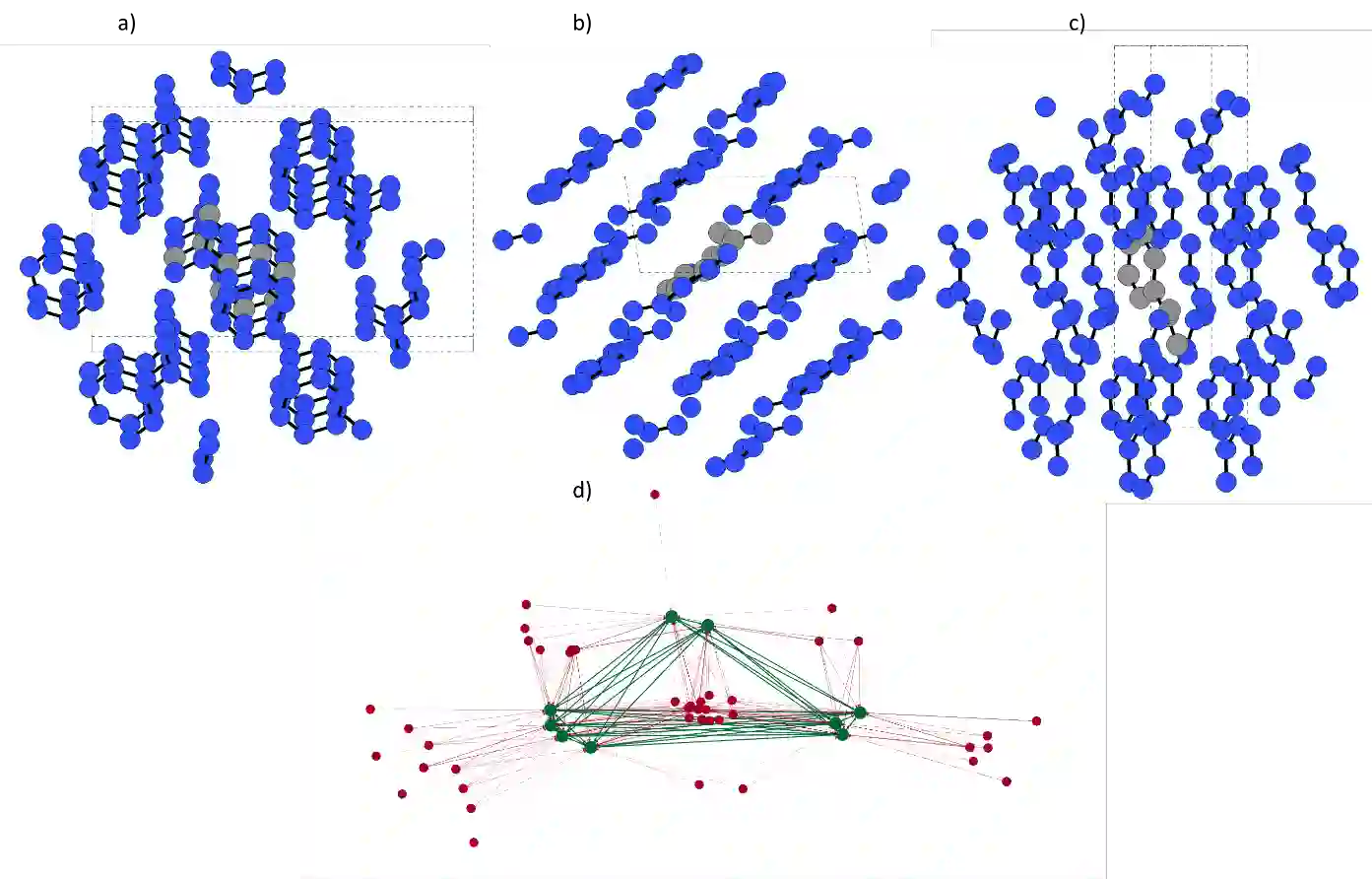
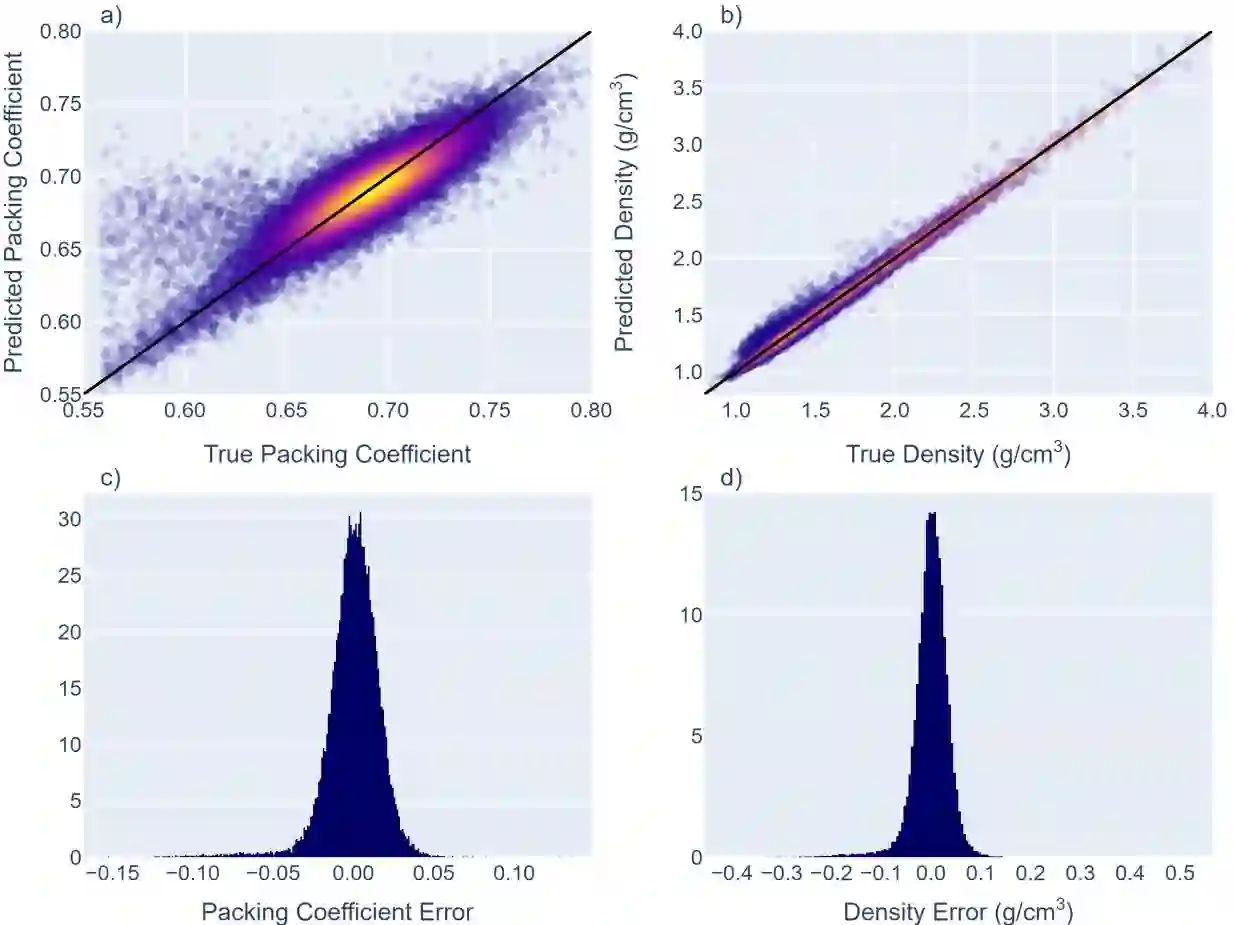
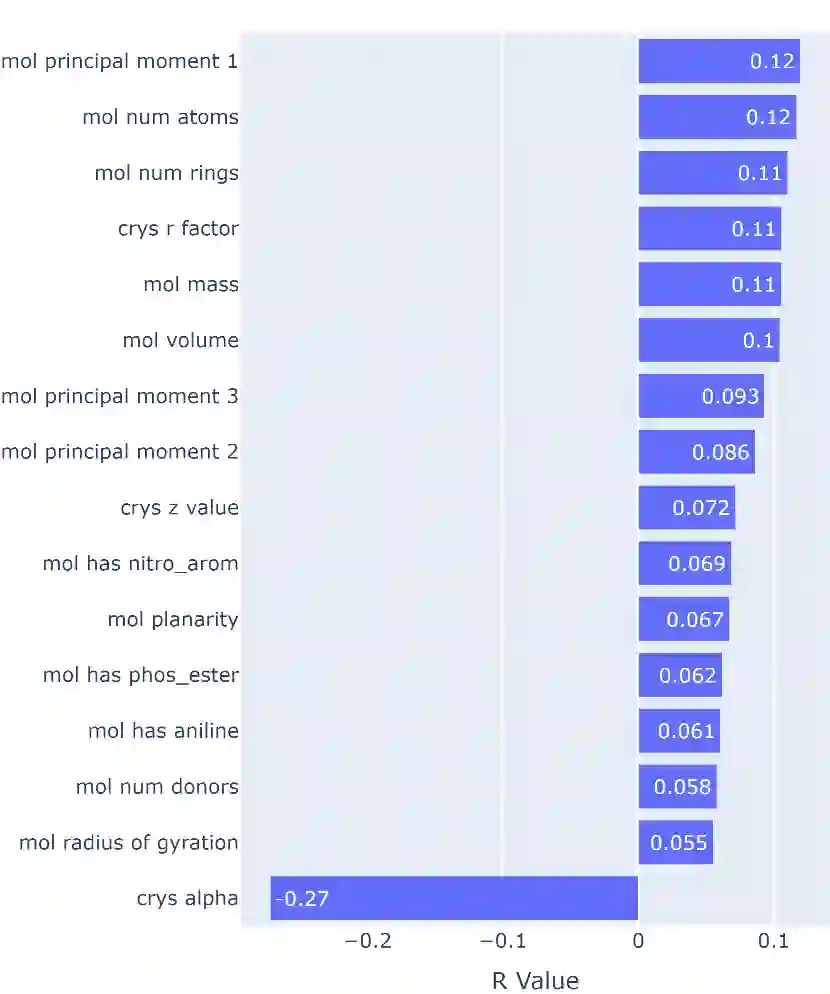
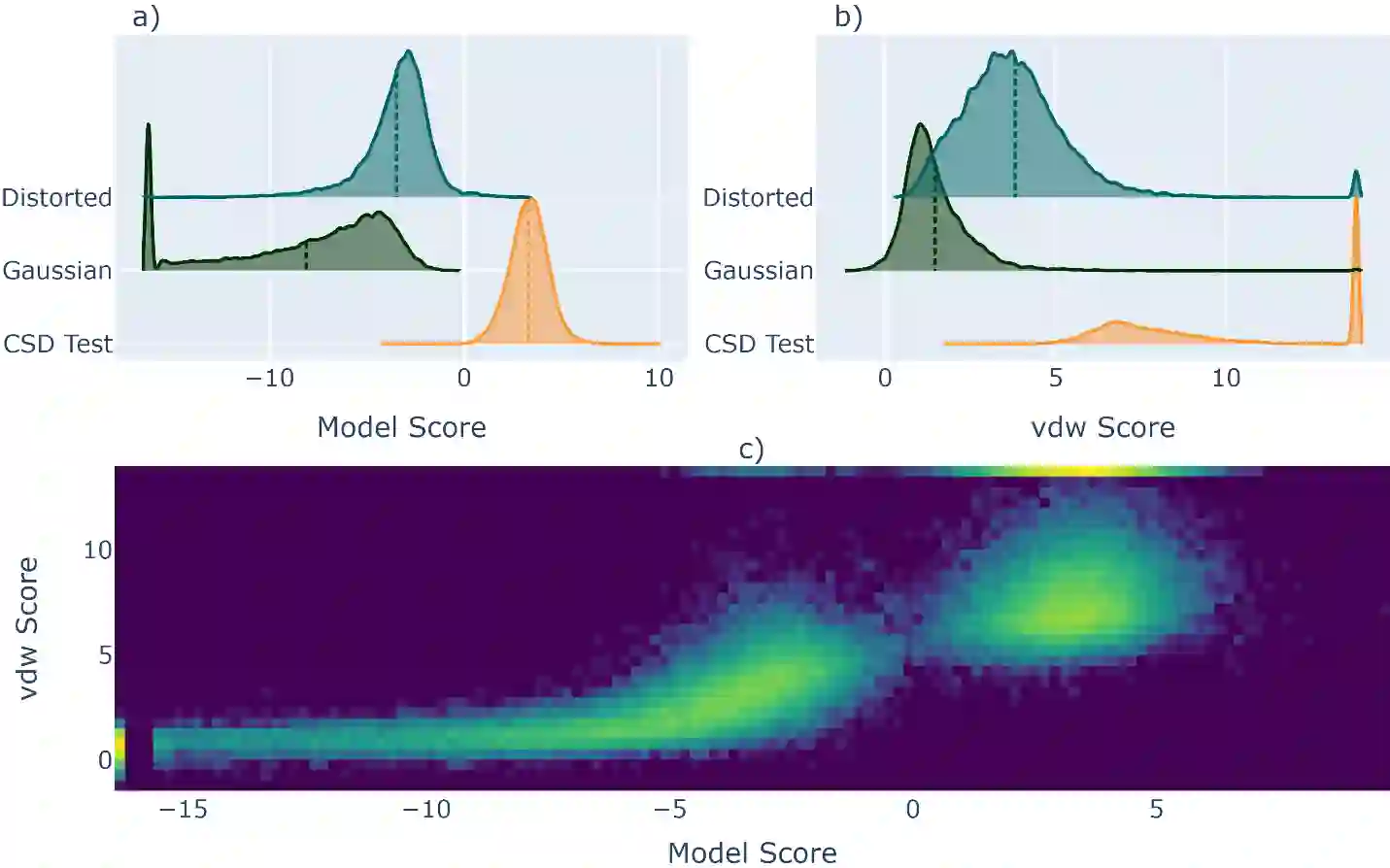
We develop and test new machine learning strategies for accelerating molecular crystal structure ranking and crystal property prediction using tools from geometric deep learning on molecular graphs. Leveraging developments in graph-based learning and the availability of large molecular crystal datasets, we train models for density prediction and stability ranking which are accurate, fast to evaluate, and applicable to molecules of widely varying size and composition. Our density prediction model, MolXtalNet-D, achieves state of the art performance, with lower than 2% mean absolute error on a large and diverse test dataset. Our crystal ranking tool, MolXtalNet-S, correctly discriminates experimental samples from synthetically generated fakes and is further validated through analysis of the submissions to the Cambridge Structural Database Blind Tests 5 and 6. Our new tools are computationally cheap and flexible enough to be deployed within an existing crystal structure prediction pipeline both to reduce the search space and score/filter crystal candidates.
翻译:我们开发并测试了新的机器学习策略,利用分子图上的几何深度学习工具,加速分子晶体结构排序与晶体性质预测。借助基于图学习的发展及大规模分子晶体数据集的可用性,我们训练了用于密度预测与稳定性排序的模型,这些模型准确、评估速度快,且适用于尺寸和成分差异显著的分子。我们的密度预测模型MolXtalNet-D在大型多样测试数据集上实现了低于2%的平均绝对误差,达到当前最优性能。晶体排序工具MolXtalNet-S能够正确区分实验样本与合成生成的假样本,并通过分析提交至剑桥结构数据库盲测5和6的结果进一步得到验证。我们的新工具计算成本低且灵活,可部署于现有晶体结构预测流程中,既能缩小搜索空间,又能对候选晶体进行评分/筛选。