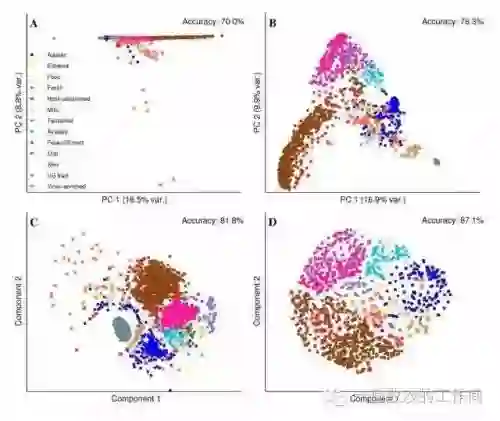
High-dimensional compositional data pose unique statistical challenges due to the simplex constraint and excess zeros. While dimension reduction is indispensable for analyzing such data, conventional approaches often rely on log-ratio transformations that compromise interpretability and distort the data through ad hoc zero replacements. To address these issues, we introduce a geometry-preserving framework for dimension reduction of compositional data, mapping high-dimensional compositions directly to a lower-dimensional simplex. This framework is interpretable as a softened amalgamation of compositions and enables dual visualization -- showing both projected data and how variables contribute to reduced components -- for at-a-glance interpretation. Within this geometry, we define a new sufficient dimension reduction (SDR) approach for compositional predictors, whose identifiable object, termed the central compositional subspace, differs from the classical central subspace in Euclidean SDR. For estimation, we propose a kernel-based method that yields sparse solutions and comes with an intrinsic predictive model for direct downstream analyses. We prove consistency through a new subspace-comparison argument that allows the estimated and target subspaces to have different dimensions. Applications to real microbiome datasets demonstrate that our approach provides a powerful graphical exploration tool for uncovering meaningful biological patterns in high-dimensional compositional data.
翻译:高维成分数据因单形约束和过量零值带来独特的统计挑战。降维对此类数据分析不可或缺,但传统方法通常依赖对数比率变换,既损害可解释性,又通过临时零值替代扭曲数据本质。针对这些问题,我们提出一种保持几何结构的成分数据降维框架,直接将高维成分映射至低维单形。该框架可解释为成分的软化合并,支持双重可视化——同时展示投影数据与变量对降维成分的贡献——实现一目了然的解读。在此几何框架内,我们定义了针对成分预测变量的新型充分降维方法,其可识别对象——称为中心成分子空间——不同于欧几里得充分降维中的经典中心子空间。在估计方面,我们提出基于核函数的稀疏解方法,该方法自带预测模型可直接用于下游分析。通过允许估计子空间与目标子空间具有不同维度的新型子空间比较论证,我们证明了估计的一致性。实际微生物组数据集的应用表明,我们的方法为揭示高维成分数据中有意义的生物学模式提供了强大的图形化探索工具。