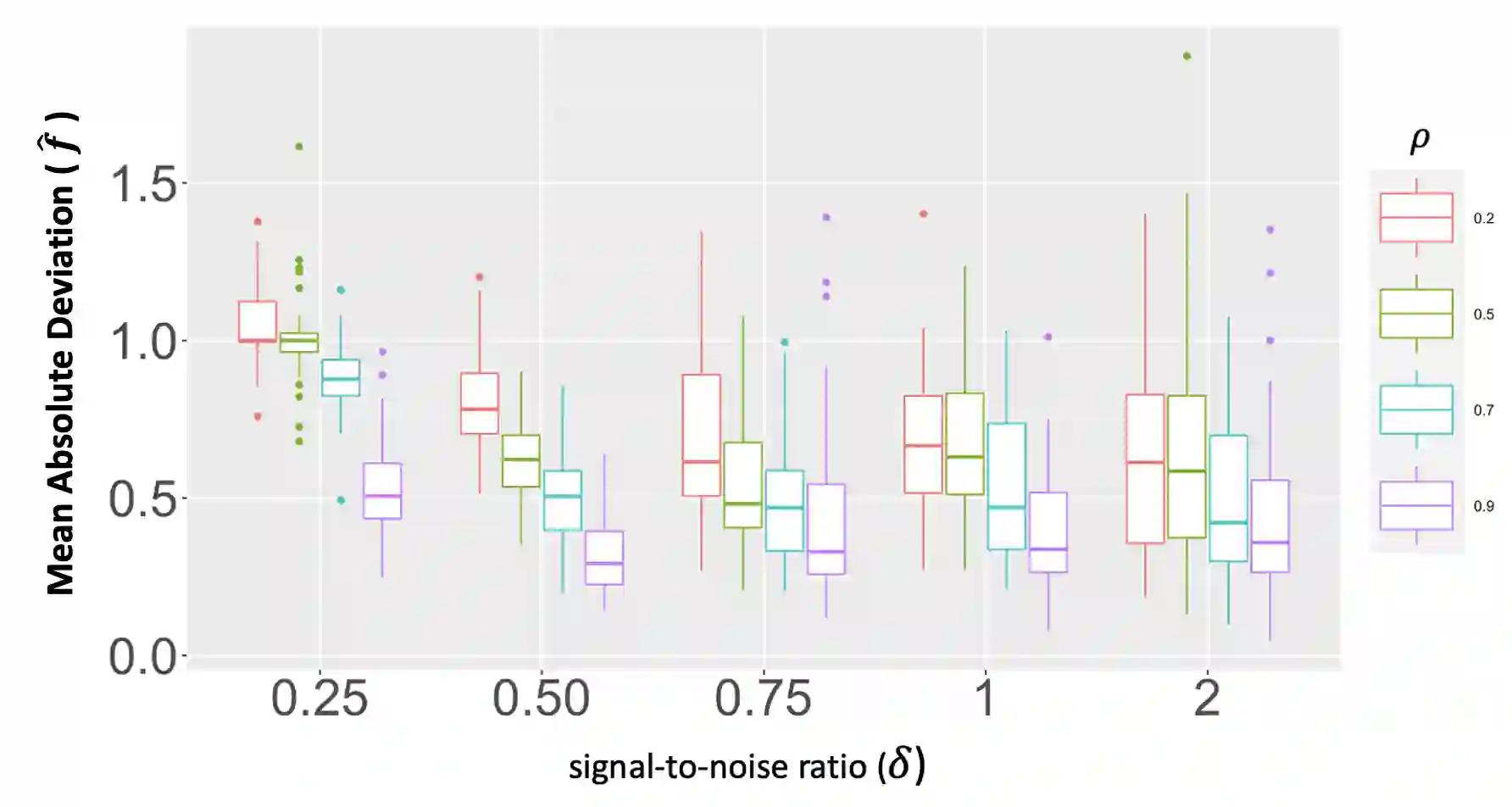
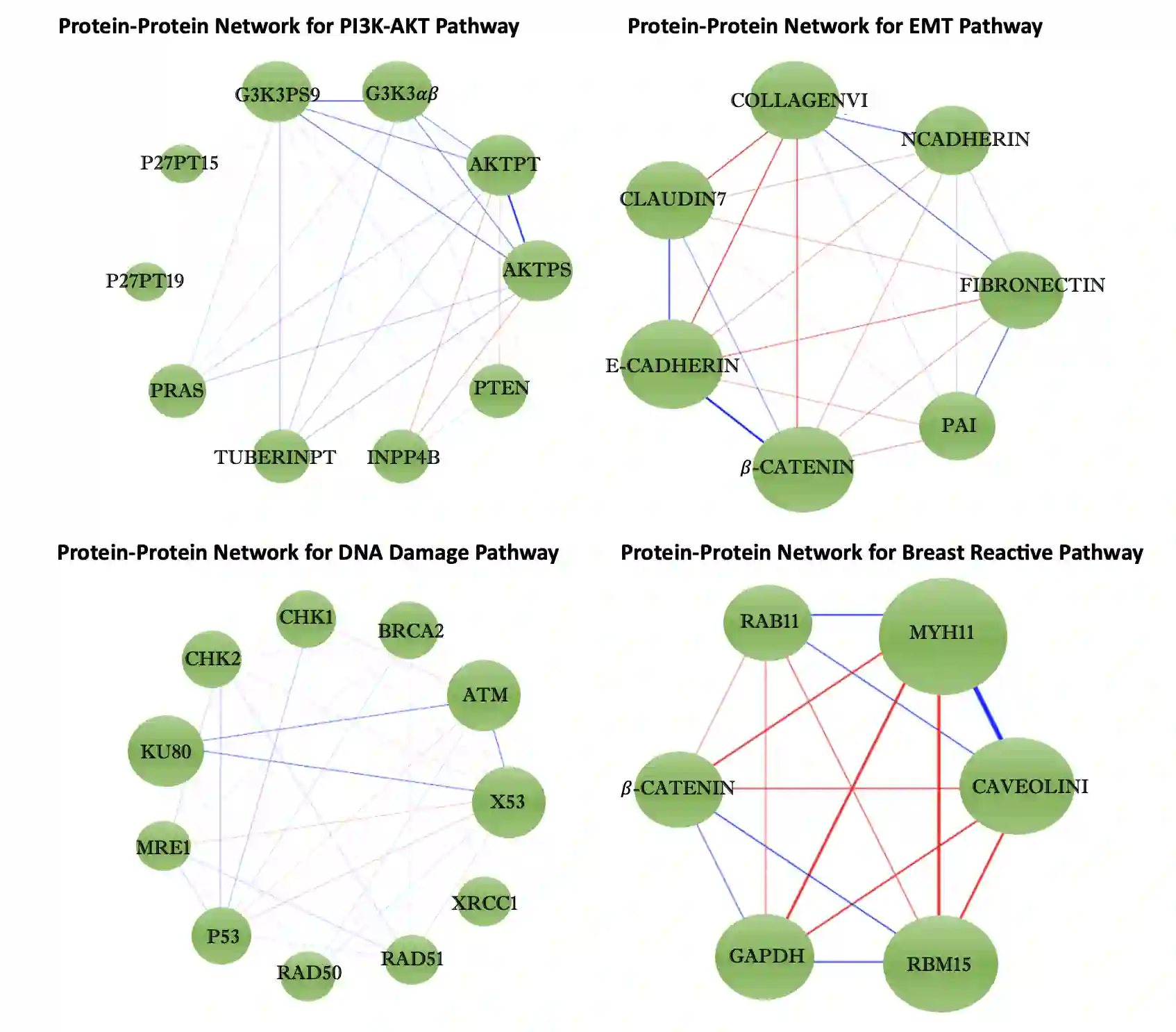
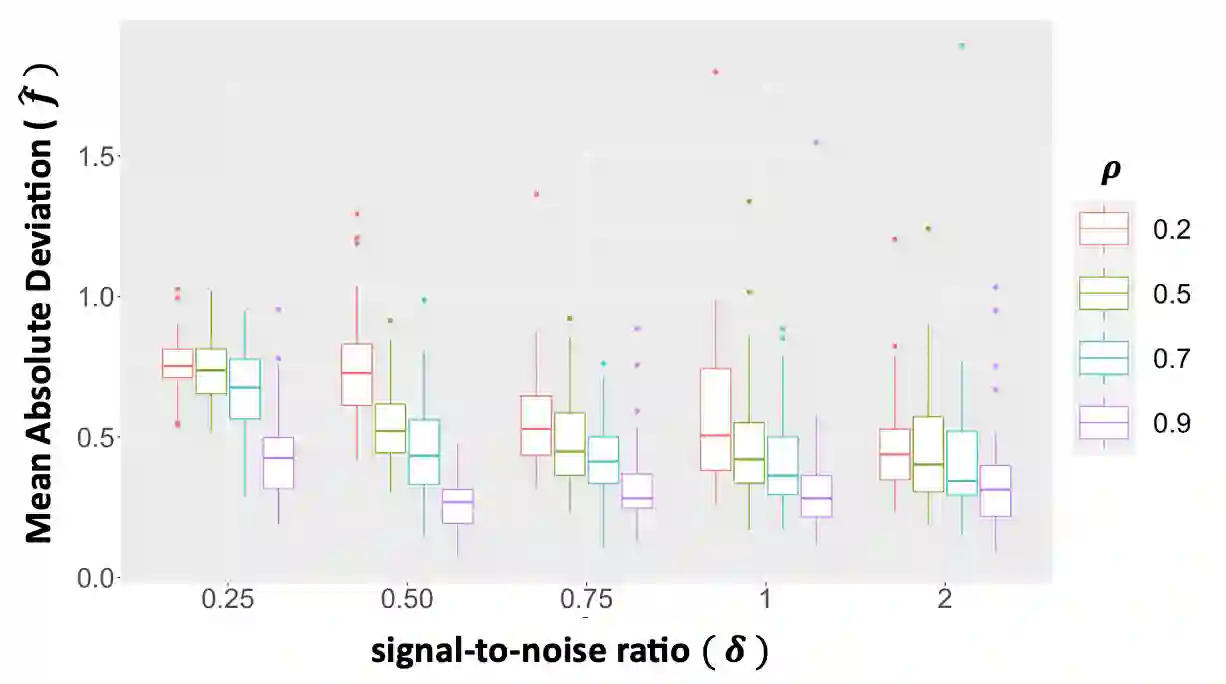
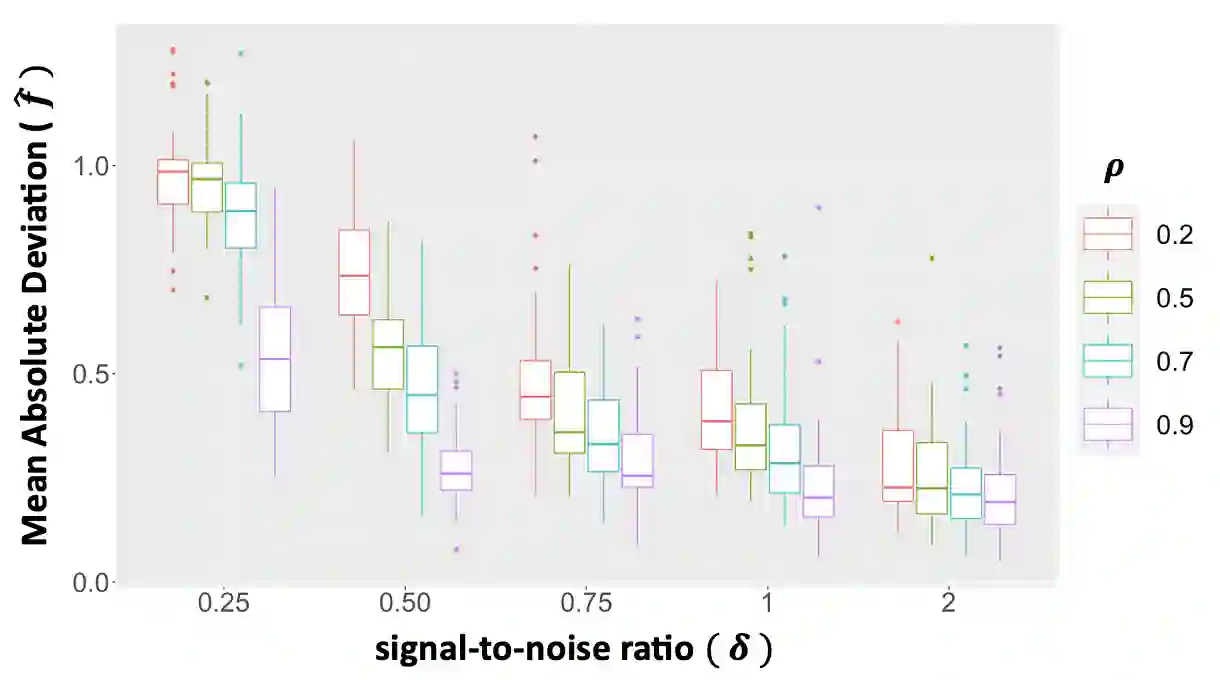
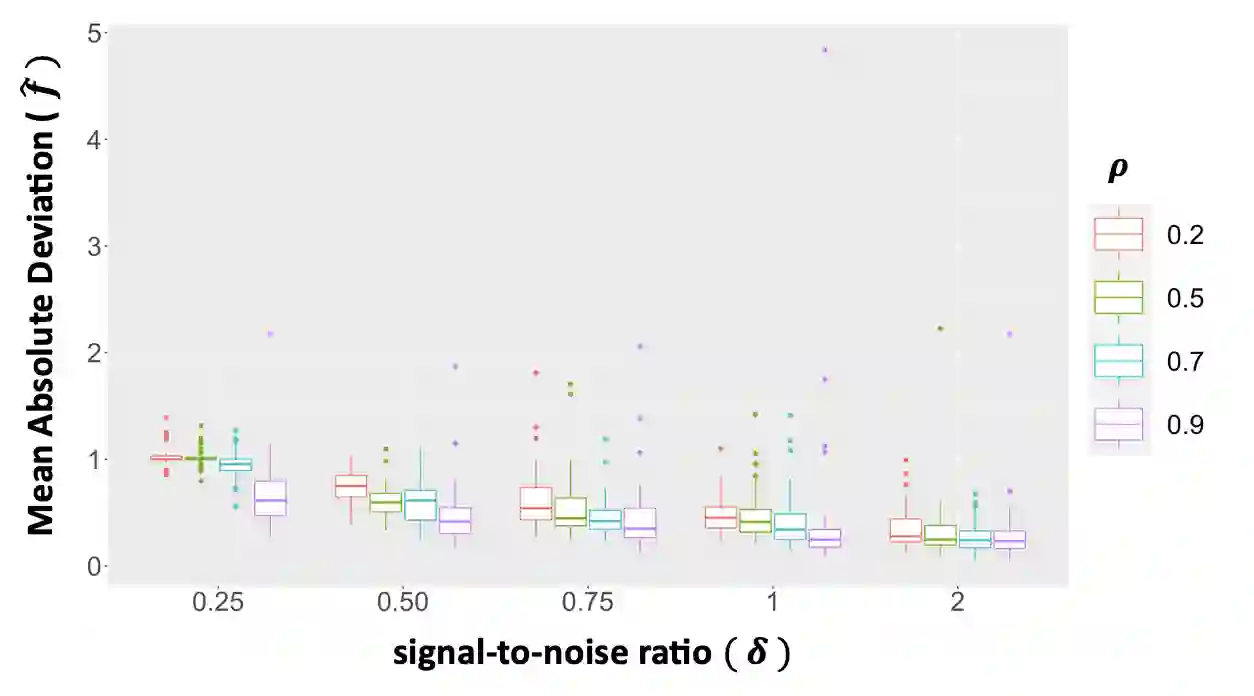
We propose a new method for the simultaneous selection and estimation of multivariate sparse additive models with correlated errors. Our method called Covariance Assisted Multivariate Penalized Additive Regression (CoMPAdRe) simultaneously selects among null, linear, and smooth non-linear effects for each predictor while incorporating joint estimation of the sparse residual structure among responses, with the motivation that accounting for inter-response correlation structure can lead to improved accuracy in variable selection and estimation efficiency. CoMPAdRe is constructed in a computationally efficient way that allows the selection and estimation of linear and non-linear covariates to be conducted in parallel across responses. Compared to single-response approaches that marginally select linear and non-linear covariate effects, we demonstrate in simulation studies that the joint multivariate modeling leads to gains in both estimation efficiency and selection accuracy, of greater magnitude in settings where signal is moderate relative to the level of noise. We apply our approach to protein-mRNA expression levels from multiple breast cancer pathways obtained from The Cancer Proteome Atlas and characterize both mRNA-protein associations and protein-protein subnetworks for each pathway. We find non-linear mRNA-protein associations for the Core Reactive, EMT, PIK-AKT, and RTK pathways.
翻译:我们提出了一种新方法,用于同时选择和估计具有相关误差的多变量稀疏加性模型。该方法名为协方差辅助的多变量惩罚加性回归(CoMPAdRe),可在为每个预测变量选择零效应、线性效应和平滑非线性效应的同时,联合估计响应变量间的稀疏残差结构。其动机在于,考虑响应间的相关结构能够提高变量选择的准确性和估计效率。CoMPAdRe采用高效的计算方式构建,允许跨不同响应并行进行线性和非线性协变量的选择与估计。与单一响应方法仅边际性地选择线性和非线性协变量效应相比,我们的模拟研究表明:联合多变量建模在估计效率和选择准确性上均有提升,尤其在信噪比中等的情况下,增益更为显著。我们将该方法应用于癌症蛋白质组图谱中多个乳腺癌通路的蛋白质-mRNA表达水平数据,刻画了每个通路的mRNA-蛋白质关联以及蛋白质-蛋白质子网络。我们在核心反应、EMT、PIK-AKT和RTK通路中发现了非线性的mRNA-蛋白质关联。