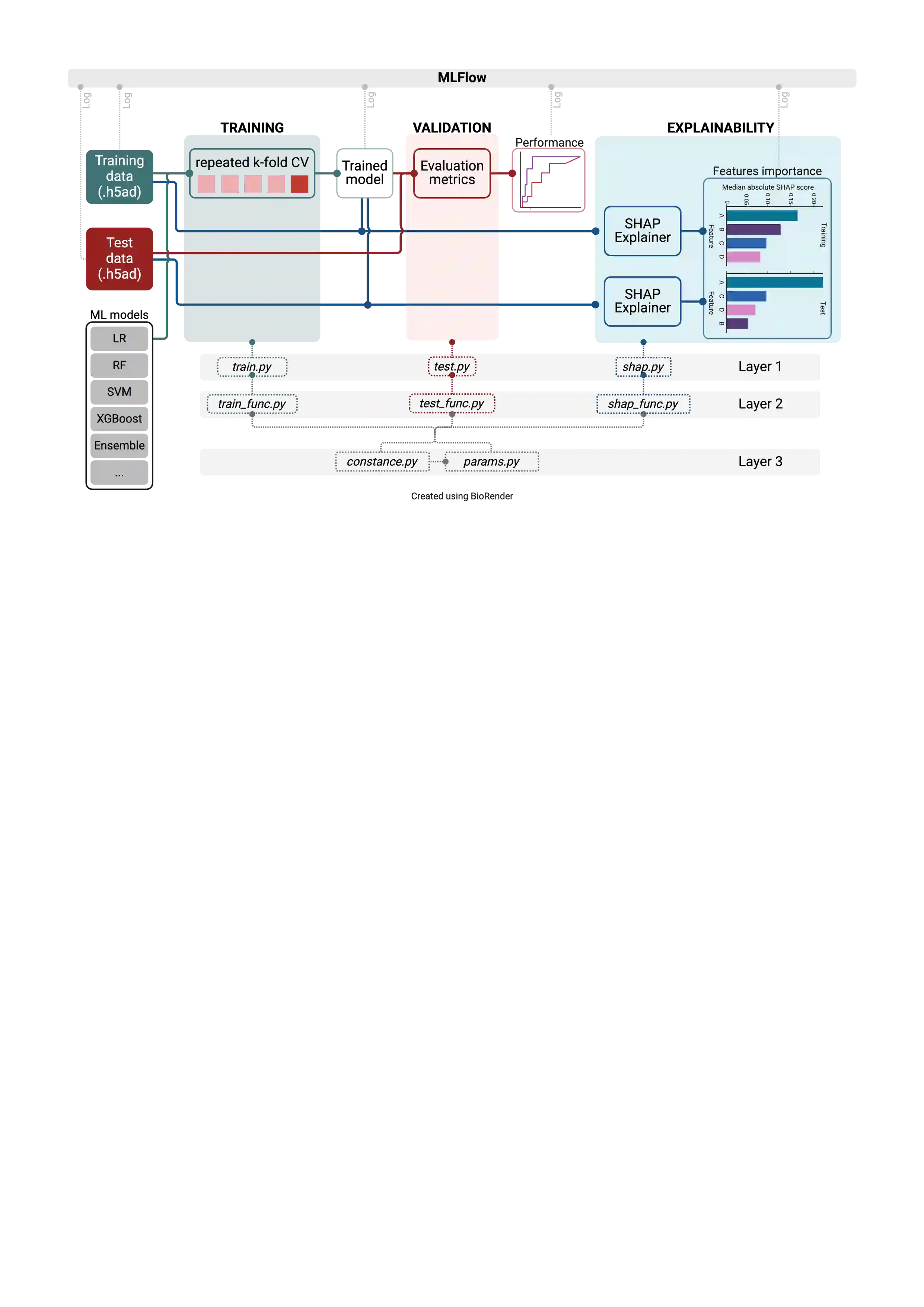
Motivation: Building and iterating machine learning models is often a resource-intensive process. In biomedical research, scientific codebases can lack scalability and are not easily transferable to work beyond what they were intended. xML-workFlow addresses this issue by providing a rapid, robust, and traceable end-to-end workflow that can be adapted to any ML project with minimal code rewriting. Results: We show a practical, end-to-end workflow that integrates scikit-learn, MLflow, and SHAP. This template significantly reduces the time and effort required to build and iterate on ML models, addressing the common challenges of scalability and reproducibility in biomedical research. Adapting our template may save bioinformaticians time in development and enables biomedical researchers to deploy ML projects. Availability and implementation: xML-workFlow is available at https://github.com/MedicalGenomicsLab/xML-workFlow.
翻译:动机:构建和迭代机器学习模型通常是一个资源密集型过程。在生物医学研究中,科学代码库可能缺乏可扩展性,且不易迁移到其原始用途之外的工作中。xML-workFlow通过提供一个快速、稳健且可追溯的端到端工作流来解决这一问题,该工作流只需极少的代码重写即可适配任何机器学习项目。结果:我们展示了一个集成scikit-learn、MLflow和SHAP的实用端到端工作流。该模板显著减少了构建和迭代机器学习模型所需的时间和精力,解决了生物医学研究中常见的可扩展性和可重复性挑战。采用我们的模板可为生物信息学家节省开发时间,并使生物医学研究人员能够部署机器学习项目。可用性与实现:xML-workFlow可在https://github.com/MedicalGenomicsLab/xML-workFlow获取。