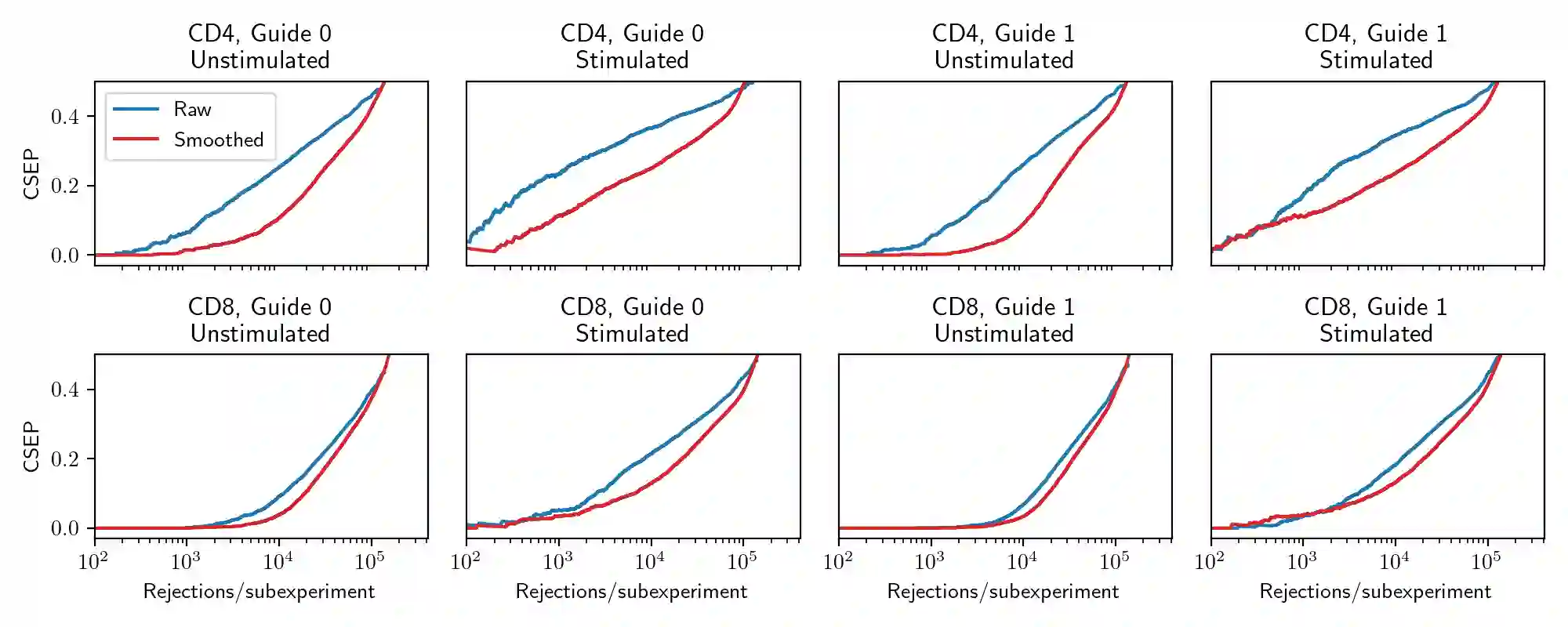
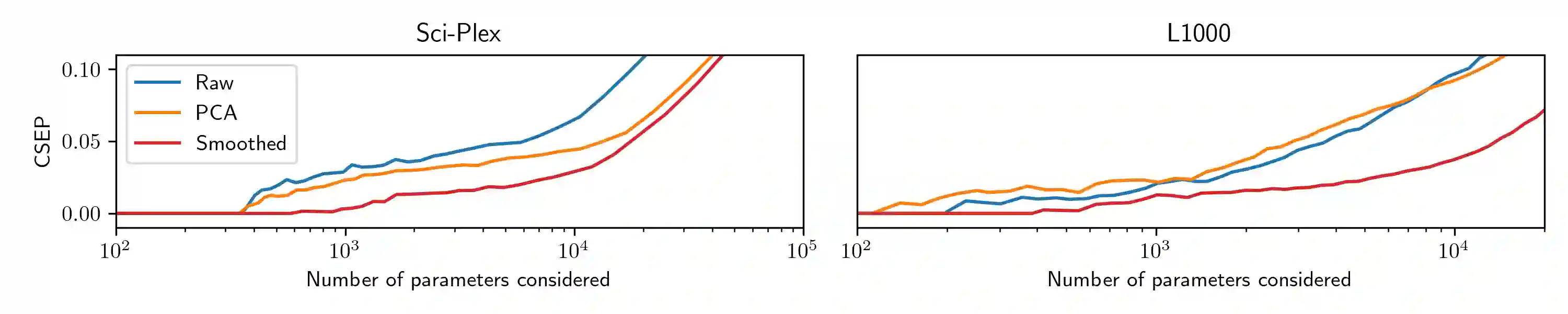
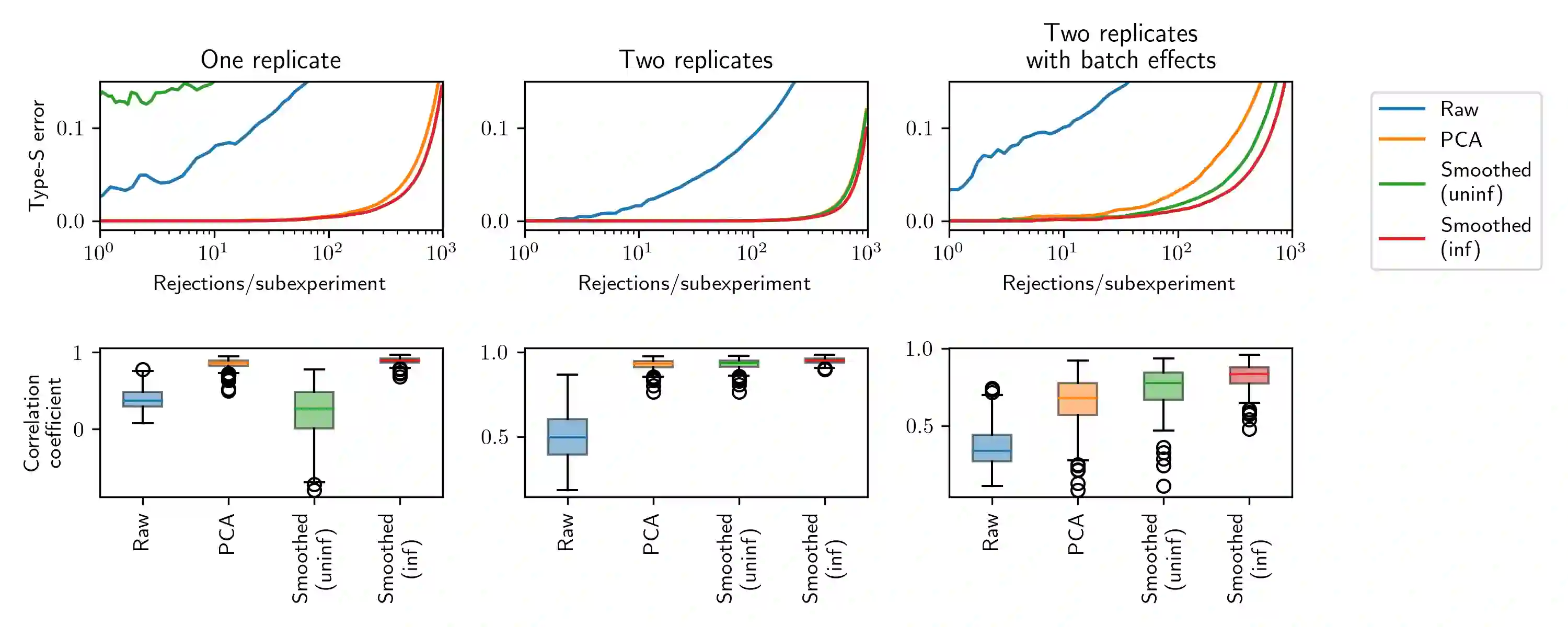
Modern cell-perturbation experiments expose cells to panels of hundreds of stimuli, such as cytokines or CRISPR guides that perform gene knockouts. These experiments are designed to investigate whether a particular gene is upregulated or downregulated by exposure to each treatment. However, due to high levels of experimental noise, typical estimators of whether a gene is up- or down-regulated make many errors. In this paper, we make two contributions. Our first contribution is a new estimator of regulatory effect that makes use of Gaussian processes and factor analysis to leverage auxiliary information about similarities among treatments, such as the chemical similarity among the drugs used to perturb cells. The new estimator typically has lower variance than unregularized estimators, which do not use auxiliary information, but higher bias. To assess whether this new estimator improves accuracy (i.e., achieves a favorable trade-off between bias and variance), we cannot simply compute its error on heldout data as ``ground truth'' about the effects of treatments is unavailable. Our second contribution is a novel data-splitting method to evaluate error rates. This data-splitting method produces valid error bounds using ``sign-valid'' estimators, which by definition have the correct sign more often than not. Using this data-splitting method, through a series of case studies we find that our new estimator, which leverages auxiliary information, can yield a three-fold reduction in type S error rate.
翻译:现代细胞扰动实验将细胞暴露于包含数百种刺激的样本中,例如细胞因子或进行基因敲除的CRISPR引导序列。这些实验旨在探究特定基因在暴露于每种处理后是否上调或下调。然而,由于实验噪声水平较高,评估基因上调或下调状态的典型估计器会产生大量错误。本文做出两项贡献。第一项贡献是提出一种新的调控效应估计器,该估计器利用高斯过程和因子分析,借助关于处理间相似性的辅助信息(例如用于扰动细胞的药物之间的化学相似性)。与不使用辅助信息的非正则化估计器相比,新估计器通常方差更低,但偏差更高。为评估该新估计器是否能提高准确性(即实现偏差与方差之间的有利权衡),我们不能简单地在留出数据上计算其误差,因为处理的效应"地面真实值"无法获取。第二项贡献是提出一种新的数据划分方法用于评估误差率。该方法利用"符号有效"估计器(其定义确保符号正确的概率大于50%)生成有效的误差界。通过这一数据划分方法,一系列案例研究表明,我们利用辅助信息的新估计器可将S类错误率降低三倍。