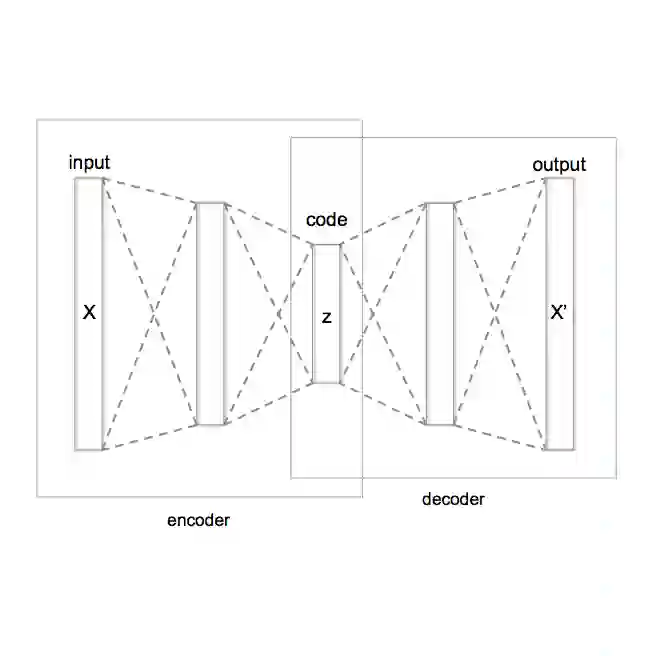
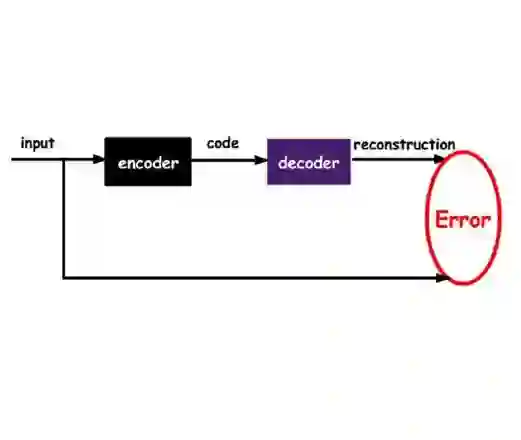
Simultaneously optimizing molecules against multiple therapeutic targets remains a profound challenge in drug discovery, particularly due to sparse rewards and conflicting design constraints. We propose a structured active learning (AL) paradigm integrating a sequence-to-sequence (Seq2Seq) variational autoencoder (VAE) into iterative loops designed to balance chemical diversity, molecular quality, and multi-target affinity. Our method alternates between expanding chemically feasible regions of latent space and progressively constraining molecules based on increasingly stringent multi-target docking thresholds. In a proof-of-concept study targeting three related coronavirus main proteases (SARS-CoV-2, SARS-CoV, MERS-CoV), our approach efficiently generated a structurally diverse set of pan-inhibitor candidates. We demonstrate that careful timing and strategic placement of chemical filters within this active learning pipeline markedly enhance exploration of beneficial chemical space, transforming the sparse-reward, multi-objective drug design problem into an accessible computational task. Our framework thus provides a generalizable roadmap for efficiently navigating complex polypharmacological landscapes.
翻译:在药物发现中,同时针对多个治疗靶点优化分子仍然是一个严峻的挑战,这尤其源于稀疏的奖励信号和相互冲突的设计约束。我们提出了一种结构化的主动学习范式,将序列到序列变分自编码器整合到迭代循环中,旨在平衡化学多样性、分子质量与多靶点亲和力。我们的方法交替进行:一方面扩展潜在空间中化学可行的区域,另一方面基于日益严格的多靶点对接阈值逐步约束分子。在一项针对三种相关冠状病毒主蛋白酶(SARS-CoV-2、SARS-CoV、MERS-CoV)的概念验证研究中,我们的方法高效地生成了一组结构多样化的泛抑制剂候选分子。我们证明,在此主动学习流程中精心安排时机并策略性地放置化学过滤器,能够显著增强对有益化学空间的探索,从而将稀疏奖励、多目标的药物设计问题转化为一项可处理的计算任务。因此,我们的框架为高效探索复杂的多药理景观提供了一条可推广的路线图。