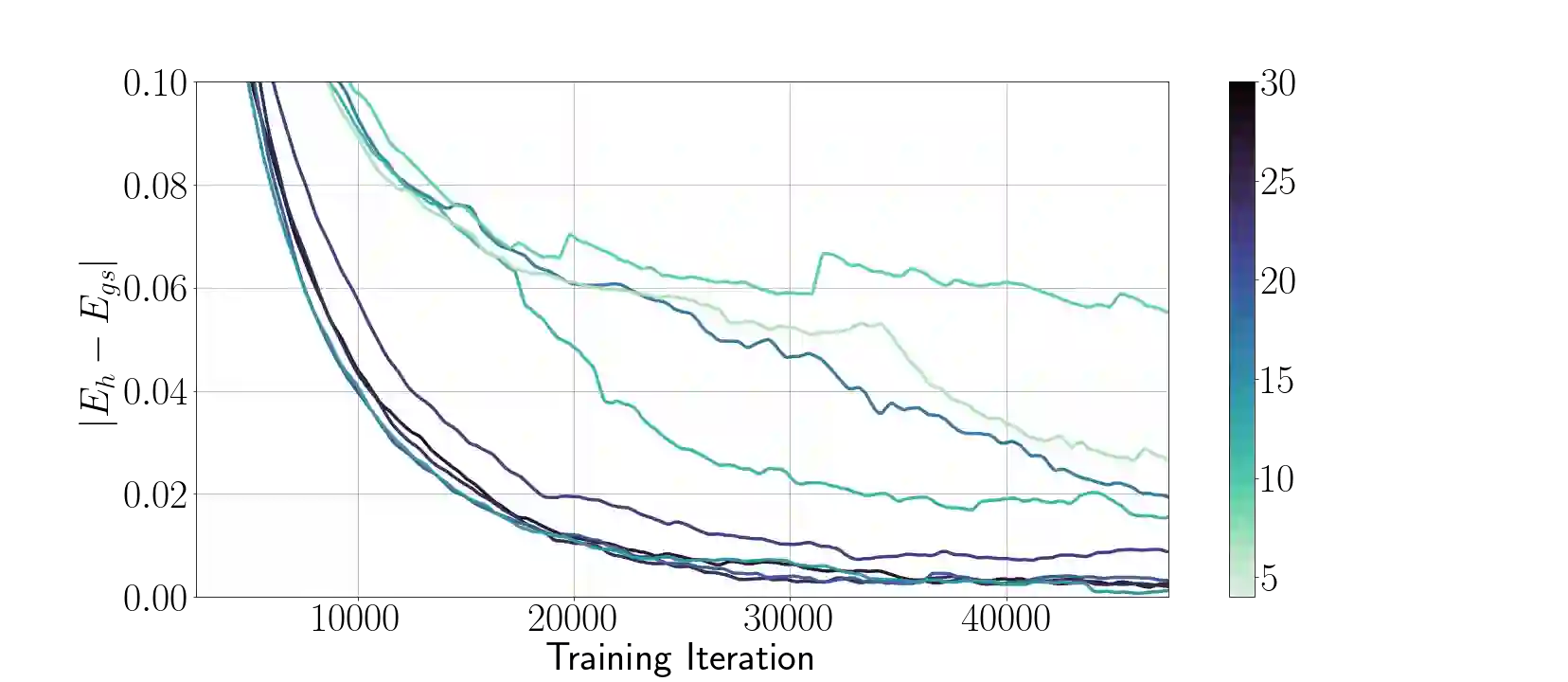
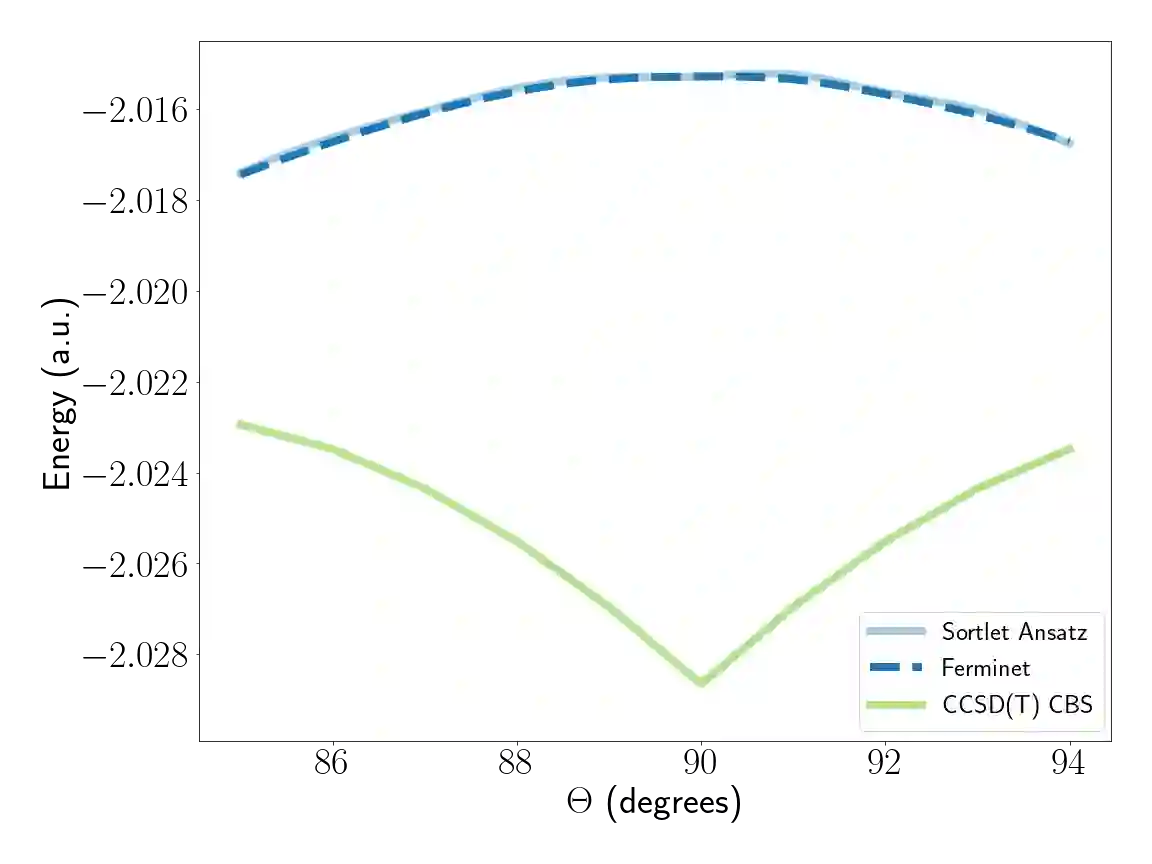
Molecular modeling at the quantum level requires choosing a parameterization of the wavefunction that both respects the required particle symmetries, and is scalable to systems of many particles. For the simulation of fermions, valid parameterizations must be antisymmetric with respect to the exchange of particles. Typically, antisymmetry is enforced by leveraging the anti-symmetry of determinants with respect to the exchange of matrix rows, but this involves computing a full determinant each time the wavefunction is evaluated. Instead, we introduce a new antisymmetrization layer derived from sorting, the $\textit{sortlet}$, which scales as $O(N \log N)$ with regards to the number of particles -- in contrast to $O(N^3)$ for the determinant. We show numerically that applying this anti-symmeterization layer on top of an attention based neural-network backbone yields a flexible wavefunction parameterization capable of reaching chemical accuracy when approximating the ground state of first-row atoms and small molecules.
翻译:量子层面的分子模拟需要选择一种既尊重所需粒子对称性,又可扩展至多粒子系统的波函数参数化方法。对于费米子模拟,有效的参数化必须对粒子交换具有反对称性。通常,反对称性是通过利用行列式对矩阵行交换的反对称性来实现的,但这需要在每次评估波函数时计算一个完整的行列式。相反,我们引入了一种基于排序的新型反对称化层——$\textit{sortlet}$,其计算复杂度随粒子数呈$O(N \log N)$扩展——相比之下,行列式为$O(N^3)$。我们通过数值实验表明,在基于注意机制的神经网络主干上应用该反对称化层,可得到灵活的波函数参数化,在近似第一行原子和小分子基态时能够达到化学精度。