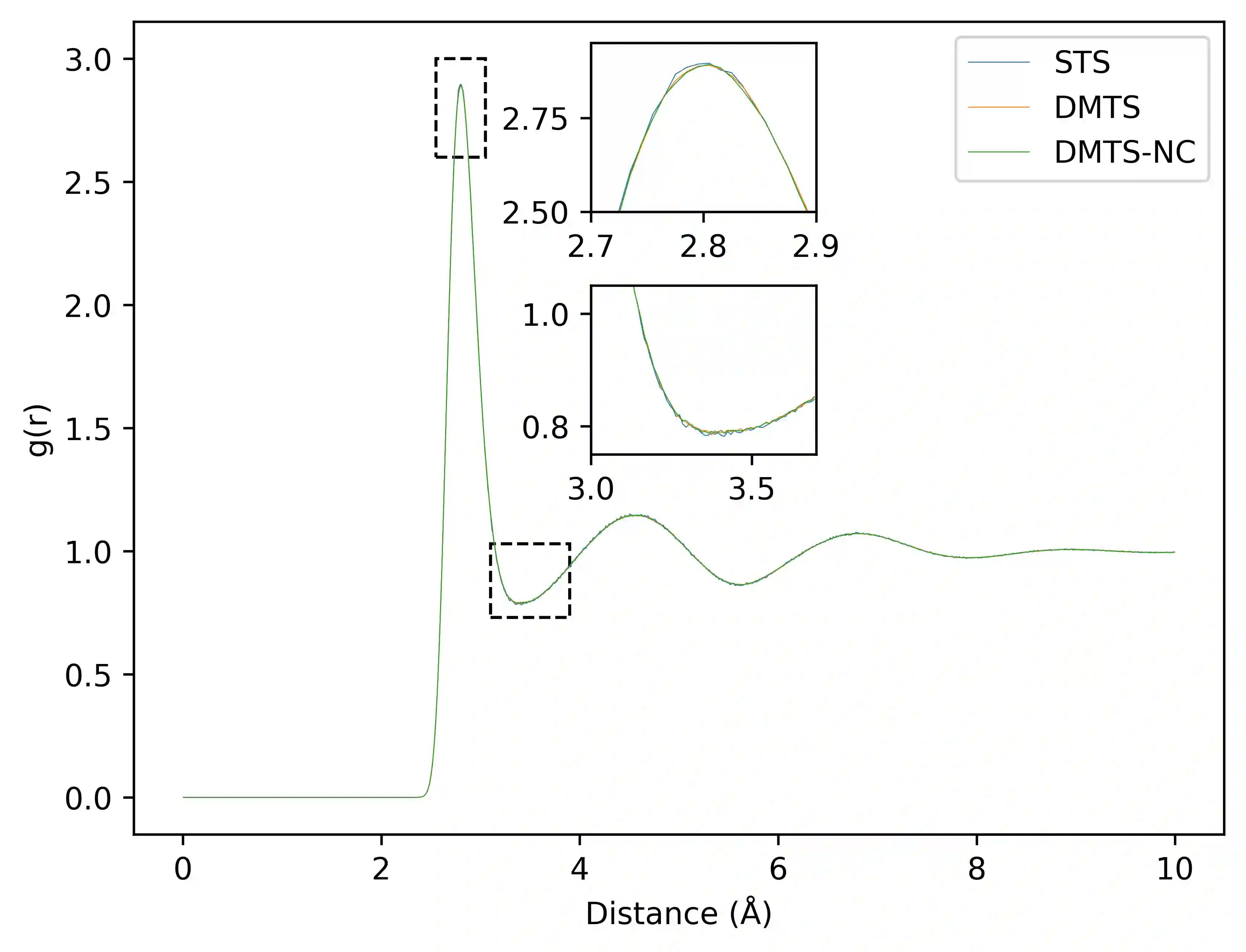
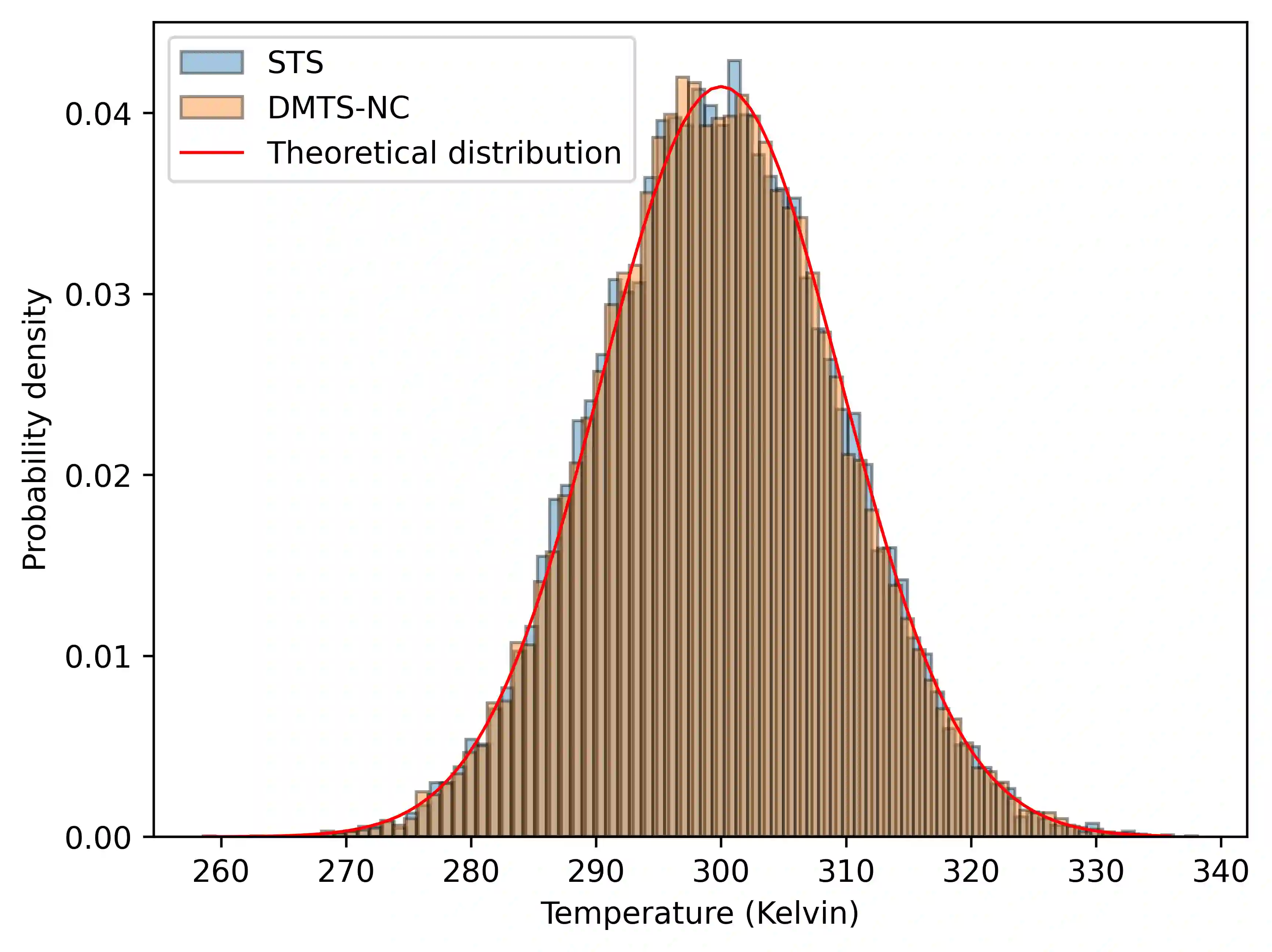
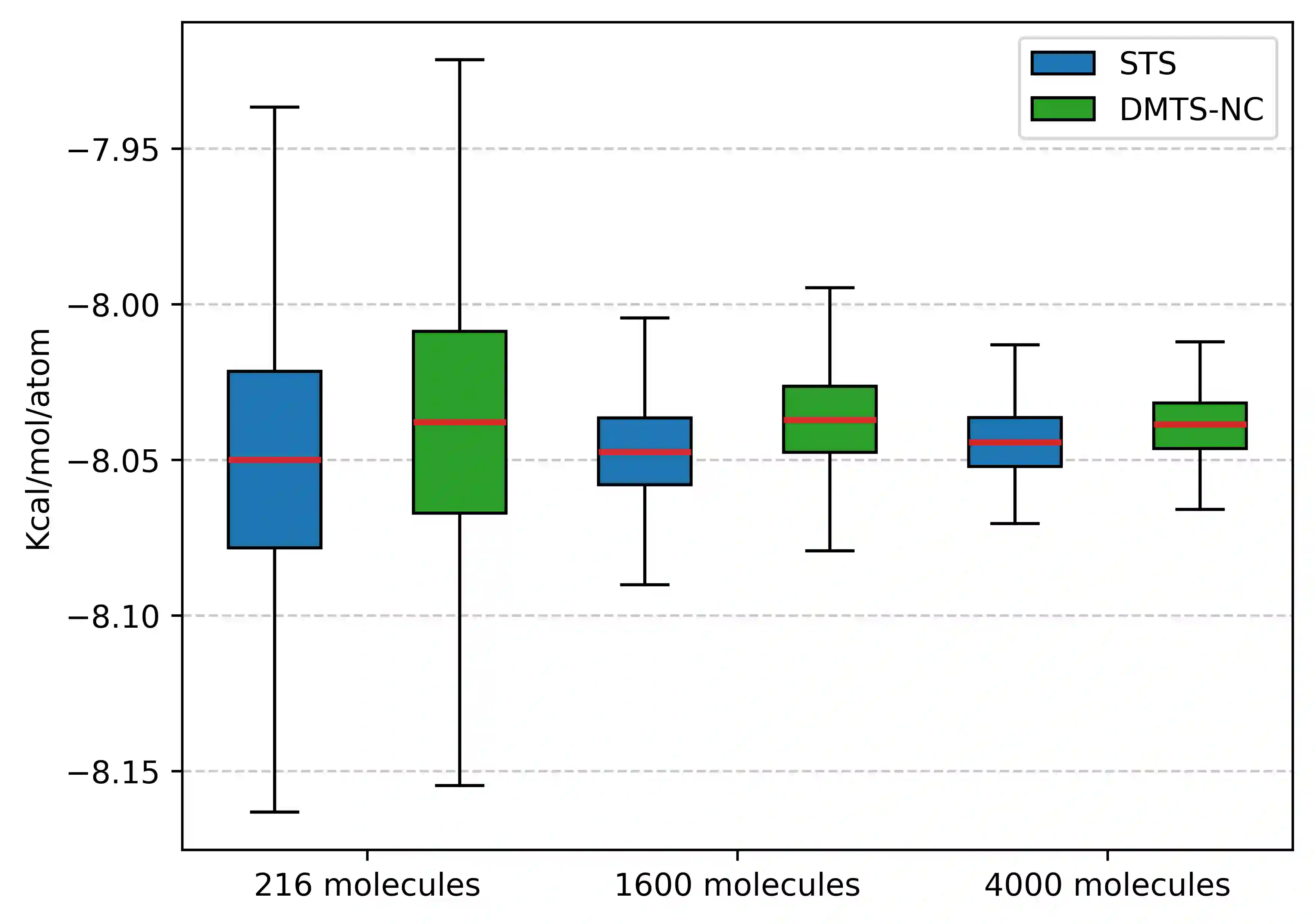
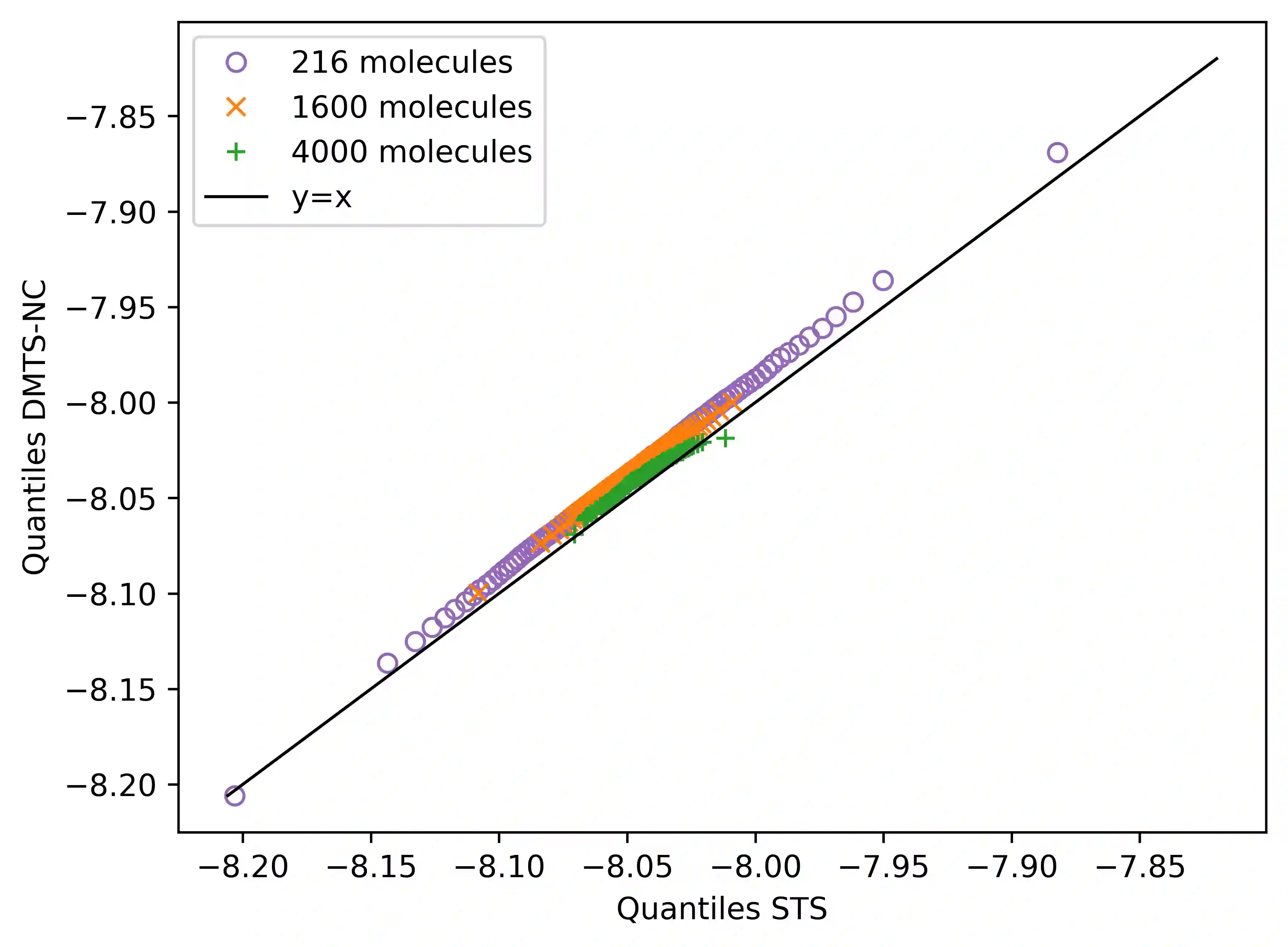
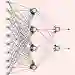
Following our previous work (J. Phys. Chem. Lett., 2026, 17, 5, 1288-1295), we propose the DMTS-NC approach, a distilled multi-time-step (DMTS) strategy using non conservative (NC) forces to further accelerate atomistic molecular dynamics simulations using foundation neural network models. There, a dual-level reversible reference system propagator algorithm (RESPA) formalism couples a target accurate conservative potential to a simplified distilled representation optimized for the production of non-conservative forces. Despite being non-conservative, the distilled architecture is designed to enforce key physical priors, such as equivariance under rotation and cancellation of atomic force components. These choices facilitate the distillation process and therefore improve drastically the robustness of simulation, significantly limiting the "holes" in the simpler potential, thus achieving excellent agreement with the forces data. Overall, the DMTS-NC scheme is found to be more stable and efficient than its conservative counterpart with additional speedups reaching 15-30% over DMTS. Requiring no finetuning steps, it is easier to implement and can be pushed to the limit of the systems physical resonances to maintain accuracy while providing maximum efficiency. As for DMTS, DMTS-NC is applicable to any neural network potential.
翻译:基于我们先前的工作(J. Phys. Chem. Lett., 2026, 17, 5, 1288-1295),本文提出DMTS-NC方法,这是一种利用非保守力进行蒸馏多时间步的策略,旨在进一步加速基于基础神经网络模型的原子分子动力学模拟。该方法采用双级可逆参考系统传播子算法形式体系,将目标精确的保守势与一个经过优化、用于生成非保守力的简化蒸馏表示相耦合。尽管是非保守的,该蒸馏架构被设计为强制满足关键的物理先验,例如旋转等变性及原子力分量的抵消。这些选择促进了蒸馏过程,从而极大提升了模拟的鲁棒性,显著减少了简化势中的“空洞”,因此与力数据达到了极好的一致性。总体而言,DMTS-NC方案比其保守对应方案更稳定高效,相较于DMTS实现了额外的15-30%加速。该方法无需微调步骤,更易于实现,并可推至系统物理共振的极限以保持精度,同时提供最大效率。与DMTS一样,DMTS-NC适用于任何神经网络势。