ICML 2026 | Autoregressive Boltzmann Generators:用自回归模型重塑分子平衡态采样
导读
分子模拟中的一个核心问题,是如何从热力学平衡态中高效采样。传统分子动力学(Molecular Dynamics, MD)可以给出可靠轨迹,但通常需要大量能量评估,且样本之间高度相关;Boltzmann Generator 则试图用生成模型直接产生低能、近似平衡的分子构象,再用精确似然和重要性采样校正偏差。 这篇论文提出 Autoregressive Boltzmann Generators(ArBG),把 Boltzmann Generator 从长期占主导的 flow-based 路线转向自回归建模。其关键思想是:不再依赖可逆变换来把简单先验推到复杂分子分布,而是像语言模型生成 token 一样,逐坐标、逐条件地建模分子构象分布。这样既保留了可计算似然,便于重要性采样修正,又绕开了 normalizing flow 在拓扑、可逆性和连续流似然计算上的限制。 论文还训练了一个 132M 参数的可迁移模型 Robin,能够在未见过的肽序列上零样本生成构象。实验显示,在 8 残基肽系统上,Robin 相比此前方法把零样本能量分布误差降低超过 60%。这说明自回归模型不仅可以用于语言、图像和分子生成,也可能成为分子平衡态采样的新型主干架构。
论文信息
论文标题:Autoregressive Boltzmann Generators 作者:Danyal Rehman, Charlie B. Tan, Yoshua Bengio, Avishek Joey Bose, Alexander Tong 论文链接:https://arxiv.org/abs/2606.27361 PDF:https://arxiv.org/pdf/2606.27361 领域:Machine Learning, Artificial Intelligence, Molecular Generative Modeling
背景:为什么需要 Boltzmann Generator
给定分子系统的势能函数 (E(x)),平衡态构象服从 Boltzmann 分布: [ p(x) \propto \exp(-E(x) / kT) ] 直接从这一分布采样并不容易。MD 通过模拟物理动力学逐步探索构象空间,但对于具有多个亚稳态、能垒较高或维度较大的体系,轨迹混合很慢,需要大量步数才能覆盖重要构象区域。对机器学习和药物发现来说,这意味着生成一个可用构象集合往往代价高昂。 Boltzmann Generator 的目标是训练一个生成模型 (q_\theta(x)),让它直接生成接近平衡态的样本。只要模型能给出精确似然,就可以进一步用重要性权重 (w(x)=p(x)/q_\theta(x)) 做校正,从而把“生成模型的速度”和“物理分布的准确性”结合起来。 过去的 Boltzmann Generator 大多基于 normalizing flow。flow 的优势是可以通过变量替换公式得到精确似然,但代价也很明显:模型必须保持可逆,映射通常受到拓扑约束;离散 flow 难以高效表示分离模式,连续 flow 又需要求解 ODE 来计算似然,推理和重采样成本较高。
核心思想:从可逆流转向自回归分解
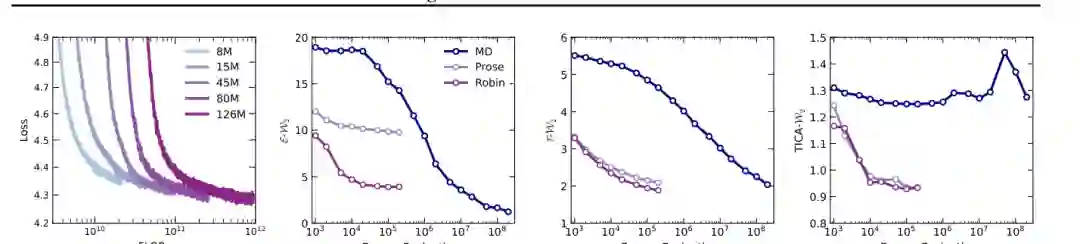
ArBG 的出发点非常直接:如果目标是既能生成样本、又能计算似然,那么自回归模型天然满足这两个条件。论文将分子构象 (x) 的联合分布分解为: [ q_\theta(x)=\prod_j q_\theta(x_j|x_{<j}) ] 其中每个坐标或离散化后的坐标分量都由前面已经生成的分量条件化预测。这样一来,模型不需要构造全局可逆映射,也不需要在连续时间流中积分;似然由一串条件概率相乘得到,和语言模型计算序列概率的方式一致。 这一改变带来三个重要优势。 第一,ArBG 不受 diffeomorphism 和拓扑保持约束限制。复杂分子分布往往由多个构象盆地构成,不同模式之间可能相隔很远。flow 在表达这类分布时容易受到连续可逆映射的结构限制,而自回归模型可以更灵活地给不同区域分配概率质量。 第二,ArBG 的似然计算更直接。连续 normalizing flow 的似然往往需要 ODE 求解和多次函数评估,而自回归模型只需一次前向或逐步条件预测即可得到 log-likelihood。这对重要性采样、重采样和序贯校正尤其关键。 第三,ArBG 可以吸收大语言模型路线中的缩放经验。论文将自回归结构扩展到不同参数规模,并在 Chignolin 十肽上展示了训练损失随计算量和模型规模改善的趋势。这意味着分子平衡态采样可能也能受益于大规模 Transformer 架构、token-level likelihood 和工程优化。
方法:ArBG 如何生成和校正分子构象
ArBG 的基本流程可以概括为四步。 首先,将分子构象表示为一组可顺序生成的变量。论文采用离散化坐标建模:把连续坐标划分到固定数量的 bins 中,模型预测每个维度属于哪个 bin,再通过 bin 内噪声重构连续坐标。这让模型可以使用成熟的分类式自回归建模,同时保留连续分子几何信息。 其次,用 Transformer 等自回归网络学习条件分布。模型在训练时最大化数据构象的似然,即让每个坐标在给定前缀条件下获得更高概率。由于每一步都是条件预测,训练目标和序列建模非常接近。 第三,生成阶段从前到后采样坐标,得到 proposal distribution 下的分子构象。ArBG 本身已经可以产生接近目标平衡态的样本,但由于它仍然是学习得到的近似分布,论文进一步引入重要性采样校正。 第四,用能量函数和模型似然计算权重,并通过 self-normalized importance sampling(SNIS)或 sequential Monte Carlo(SMC)进行重采样。直观地说,模型负责快速提出候选构象,物理能量函数负责判断这些构象在真实 Boltzmann 分布下是否合理。 这套设计的关键不是简单地“用 Transformer 生成分子”,而是把自回归似然、物理能量和重要性采样放到同一个 Boltzmann Generator 框架中。模型既要会生成,也要能被物理分布纠偏。
推理时干预:温度、重采样与 Twisted SMC
自回归模型的另一个优势,是生成过程天然分步展开,因此可以在推理时做细粒度干预。论文重点讨论了采样温度、SNIS 重采样以及 Twisted SMC 等机制。 采样温度会改变 proposal distribution 的形状。温度过低时,模型更偏向高似然、低能区域,可能导致构象模式缺失;温度过高时,生成会更分散,但也可能过度覆盖不重要区域,导致样本质量下降。论文在 AL3 上发现最优温度约为 (T=1.02),说明最佳 proposal 并不总是原始模型分布,而是需要针对能量分布和重采样目标微调。 SNIS 则利用 (p(x)/q_\theta(x)) 形式的权重重新加权样本。由于 ArBG 能提供精确 proposal likelihood,这一步可以直接实施。对于更长或更复杂系统,论文还探索了 SMC 变体,在生成过程中逐步引入目标分布信息,以减少权重退化。
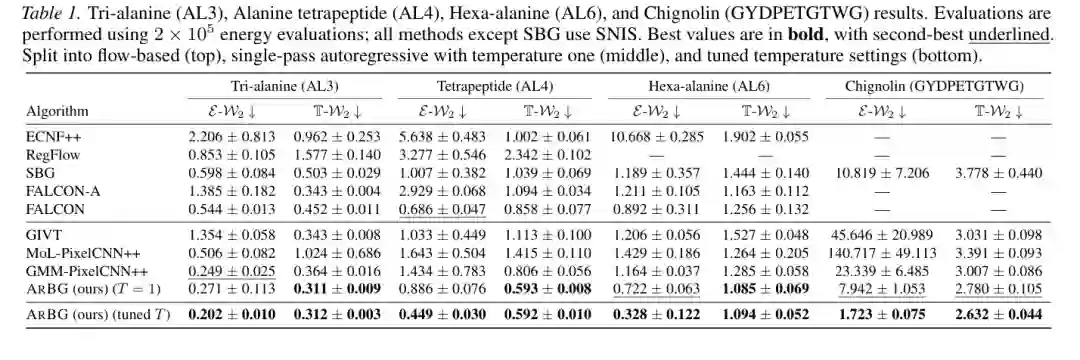
实验一:单肽系统上的表现
论文首先在 AL3、AL4、AL6 和 Chignolin 等单肽系统上评估 ArBG,并与多种 flow-based Boltzmann Generator、GMM-PixelCNN++、BG Prose、GIVT 等方法比较。评估指标主要包括:
- E-W2:能量分布的 Wasserstein-2 距离,衡量生成样本是否匹配真实能量分布。
- T-W2:扭转角分布的 Wasserstein-2 距离,衡量局部构象几何是否接近 MD 参考。
- TICA-W2:慢动力学低维表示上的分布距离,衡量生成模型是否覆盖关键构象模式。
结果显示,ArBG 在所有单肽系统上都取得了非常强的表现。尤其在更难的 AL6 和 Chignolin 上,自回归模型的优势更明显。以 Chignolin 为例,经过温度调节的 ArBG 在 (2 \times 10^5) 次能量评估预算下取得 E-W2=1.723、T-W2=2.632;相比之下,SBG 的 E-W2 为 10.819,GIVT 为 45.646,GMM-PixelCNN++ 为 23.339。也就是说,ArBG 不只是略优,而是在复杂小蛋白系统上显著缩小了与 MD 参考分布的差距。
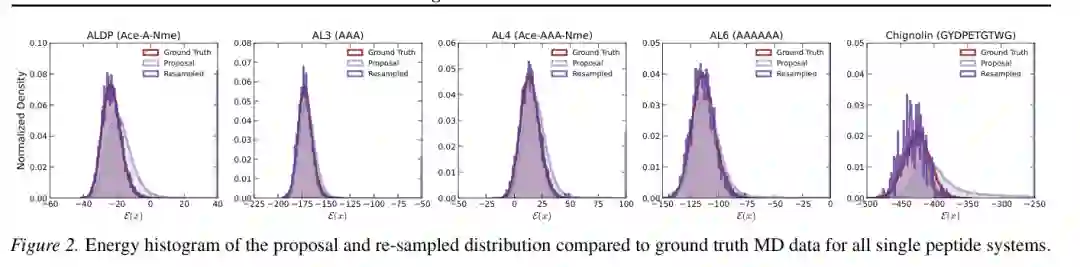
能量直方图进一步展示了这种优势。ArBG 的 proposal 分布已经覆盖主要能量区域,经过重采样后与 ground-truth MD 分布更接近。对于 Chignolin 这类分布更复杂的系统,proposal 和重采样分布之间的差异也说明,精确似然和物理重加权对于最终质量仍然非常重要。
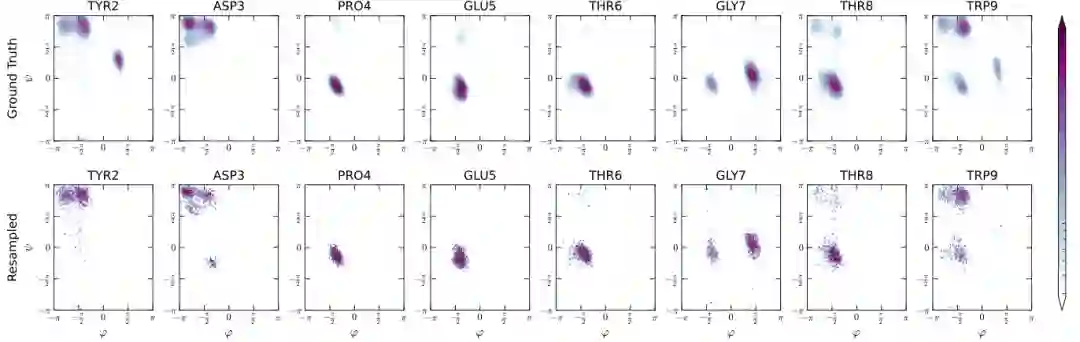
在构象空间上,论文使用 Chignolin 的 Ramachandran plots 检查各残基扭转角模式。图中上排为真实 MD 分布,下排为 ArBG 重采样结果。可以看到,ArBG 能捕捉多个残基的主要构象盆地,尤其在 PRO4、GLU5、THR6、GLY7 等局部结构上与参考分布有较高一致性。这说明模型并非只匹配了能量统计,而是学习到了有物理意义的构象模式。
实验二:Robin 可迁移模型
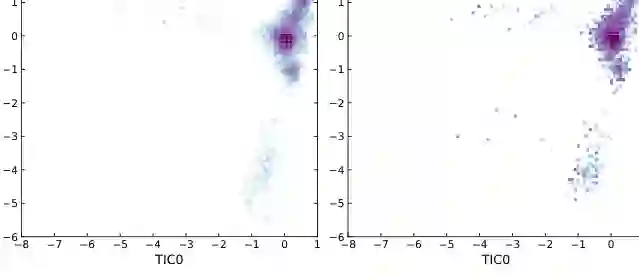
除了为单个系统训练专用生成器,论文还提出 132M 参数的可迁移模型 Robin。Robin 仍然基于 ArBG 框架,但通过序列条件化来处理不同肽序列,从而可以在未见过的分子上零样本生成构象。 这一点非常关键。传统 Boltzmann Generator 往往需要针对一个具体系统重新训练;如果每换一个肽序列都要重新训练,模型在实际应用中的扩展性会受到限制。Robin 则更接近“通用分子构象生成器”:给定新序列后,模型直接提出候选构象,再用能量评估和重要性采样校正。 在 ManyPeptidesMD 数据集上,Robin 在 4 残基和 8 残基肽系统中都取得了强结果。以 (2 \times 10^5) 次能量评估预算为例,Robin 在 8 残基系统上的 E-W2 为 3.615,而 BG Prose 为 9.360,相当于把能量分布误差降低约 61%。T-W2 和 TICA-W2 也保持竞争力,说明 Robin 不仅匹配低阶能量统计,也能更好覆盖慢变量结构。 论文还展示了 octapeptide CGSWHKQR 的 TICA 可视化。真实 MD 分布与 Robin 预测在主要慢变量区域上高度重叠,说明模型能够在零样本条件下捕捉重要构象盆地,而不是只生成局部低能样本。
消融分析:离散分辨率与采样温度
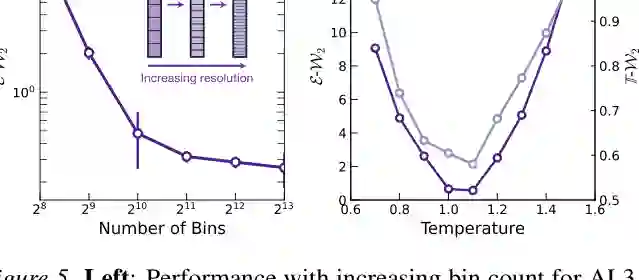
论文对两个关键设计做了消融:离散化 bin 数量和采样温度。 对于 bin 数量,结果显示分辨率越高,重采样后的 E-W2 越好。直观上,更细的 bin 能更准确表示连续坐标,减少量化误差;但它也会增加建模难度和输出空间规模。因此,bin resolution 实际上是表达精度和训练/推理复杂度之间的平衡。 对于温度,论文观察到明显的 U 形曲线:温度太低会压缩多样性,温度太高会牺牲质量。AL3 上的最优温度约为 1.02,接近但不等于 1。这一发现提示我们,Boltzmann Generator 中的最佳 proposal 不一定是最大似然训练出的原始模型分布,而是需要为后续重要性重采样服务。
与相关工作的关系
这篇论文处在三条研究线的交叉点。 第一是 Boltzmann Generator。此前工作主要围绕 normalizing flow 展开,包括离散 flow、连续 flow、能量函数辅助训练以及稳定性改进。ArBG 的贡献是把 Boltzmann Generator 的主干从可逆流换成自回归模型,从结构上绕开部分 flow bottleneck。 第二是分子生成模型。很多 Transformer 和 diffusion 模型已经用于分子结构生成,但它们通常关注有效构象、分子设计或条件生成,并不一定提供可用于 Boltzmann 校正的精确 proposal likelihood。ArBG 的重点是 equilibrium sampling,而不是只生成看起来合理的分子。 第三是自回归连续空间建模。PixelCNN、mixture density networks 和现代自回归图像/序列模型都证明了条件分解的表达能力。ArBG 将这种能力引入分子平衡态采样,并强调似然、能量和重采样三者的闭环。
总结
Autoregressive Boltzmann Generators 的意义在于,它重新定义了 Boltzmann Generator 的可选架构。过去这一方向几乎默认要使用 normalizing flow,因为 flow 能生成样本并计算似然;但这篇论文表明,自回归模型同样可以满足这两个条件,而且在表达复杂多峰分子分布、扩展到大模型和做推理时干预方面更自然。 从实验看,ArBG 在单肽系统上显著优于多种基线,在 Chignolin 等更复杂系统上优势尤其明显;Robin 则进一步展示了零样本迁移能力,使 Boltzmann Generator 从“每个系统单独训练”迈向“可迁移分子采样模型”。如果这一方向继续扩展到更大蛋白、配体-蛋白体系和更真实的溶剂环境,自回归 Boltzmann Generator 有望成为连接大模型生成能力与物理平衡态采样的重要路线。