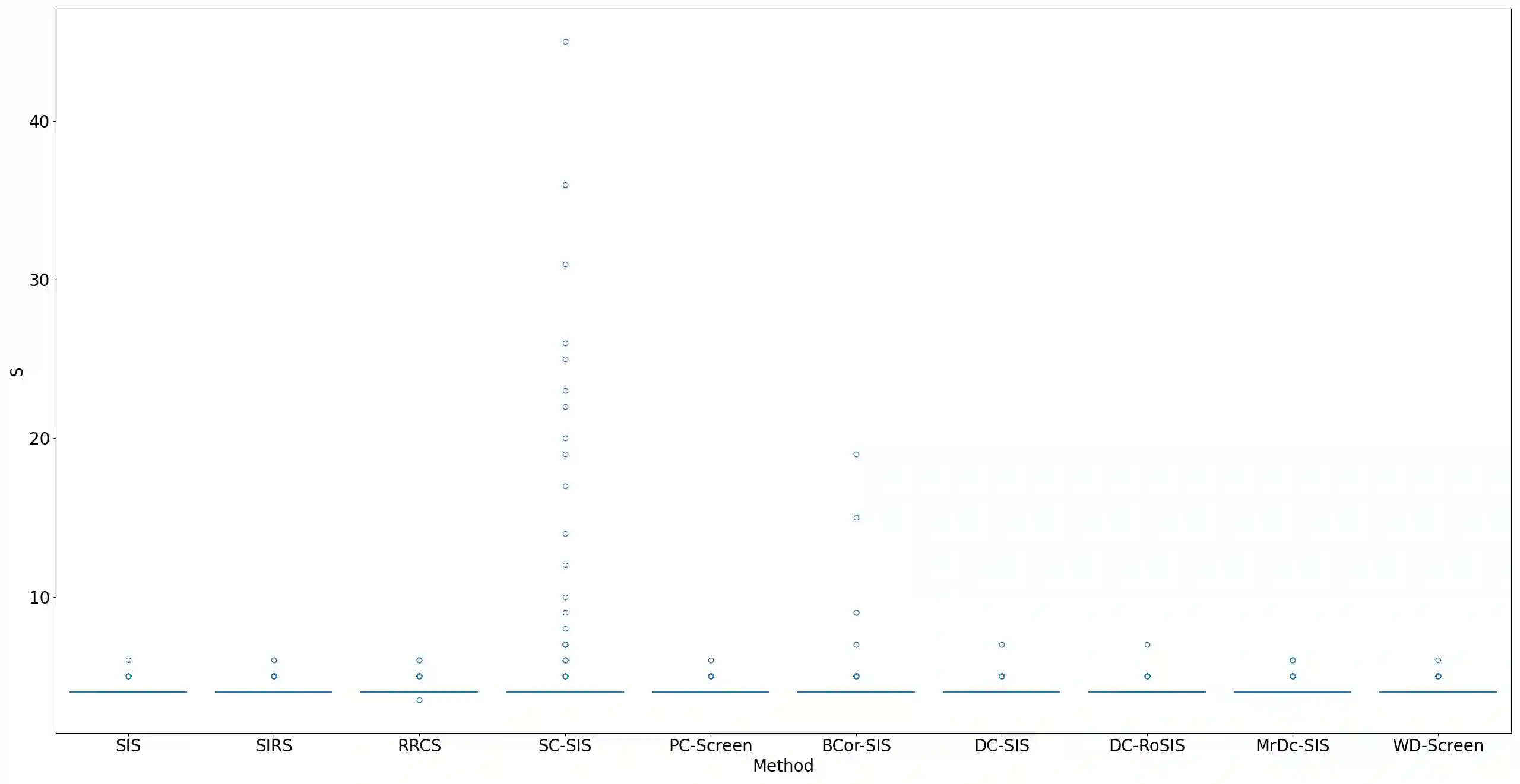
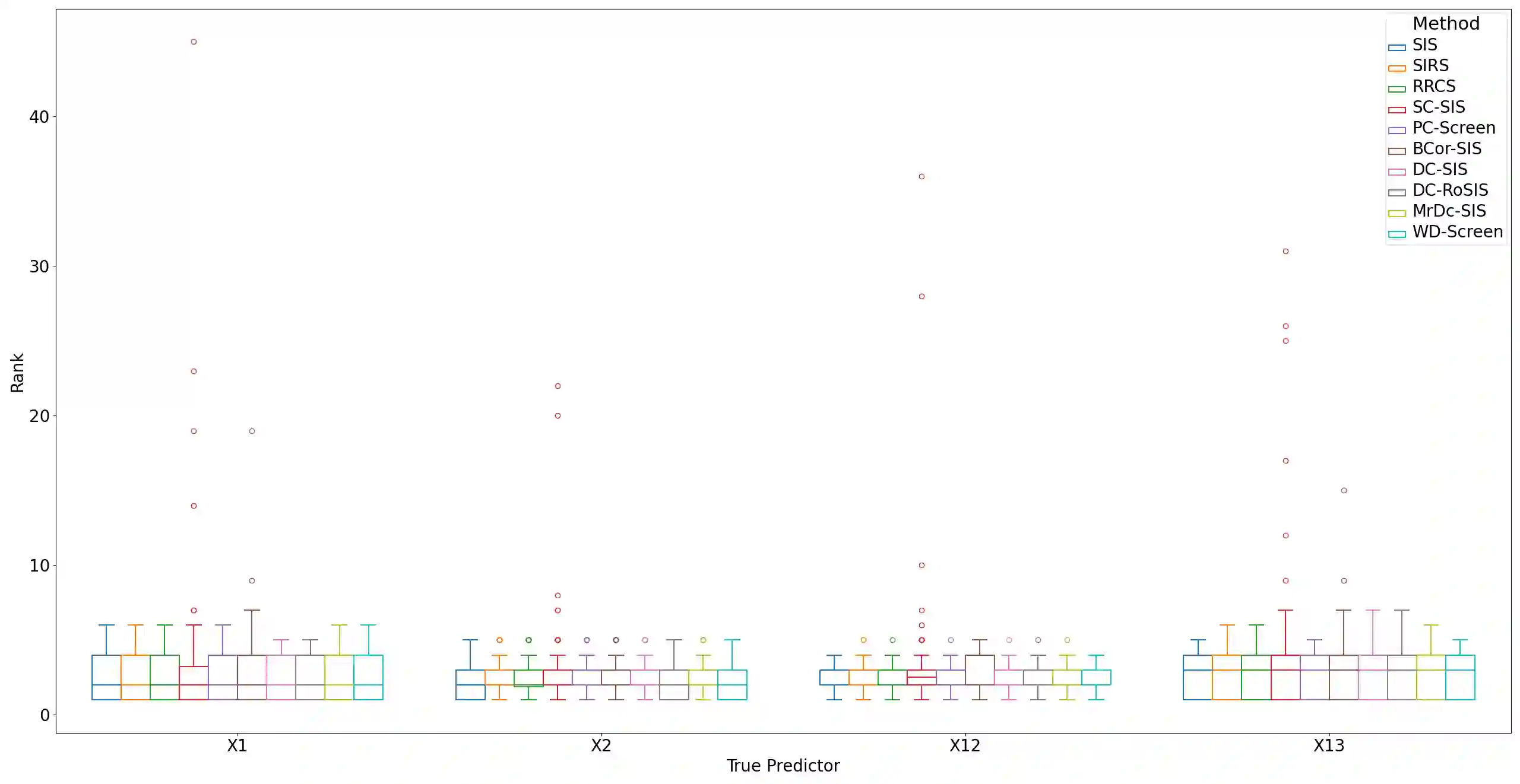
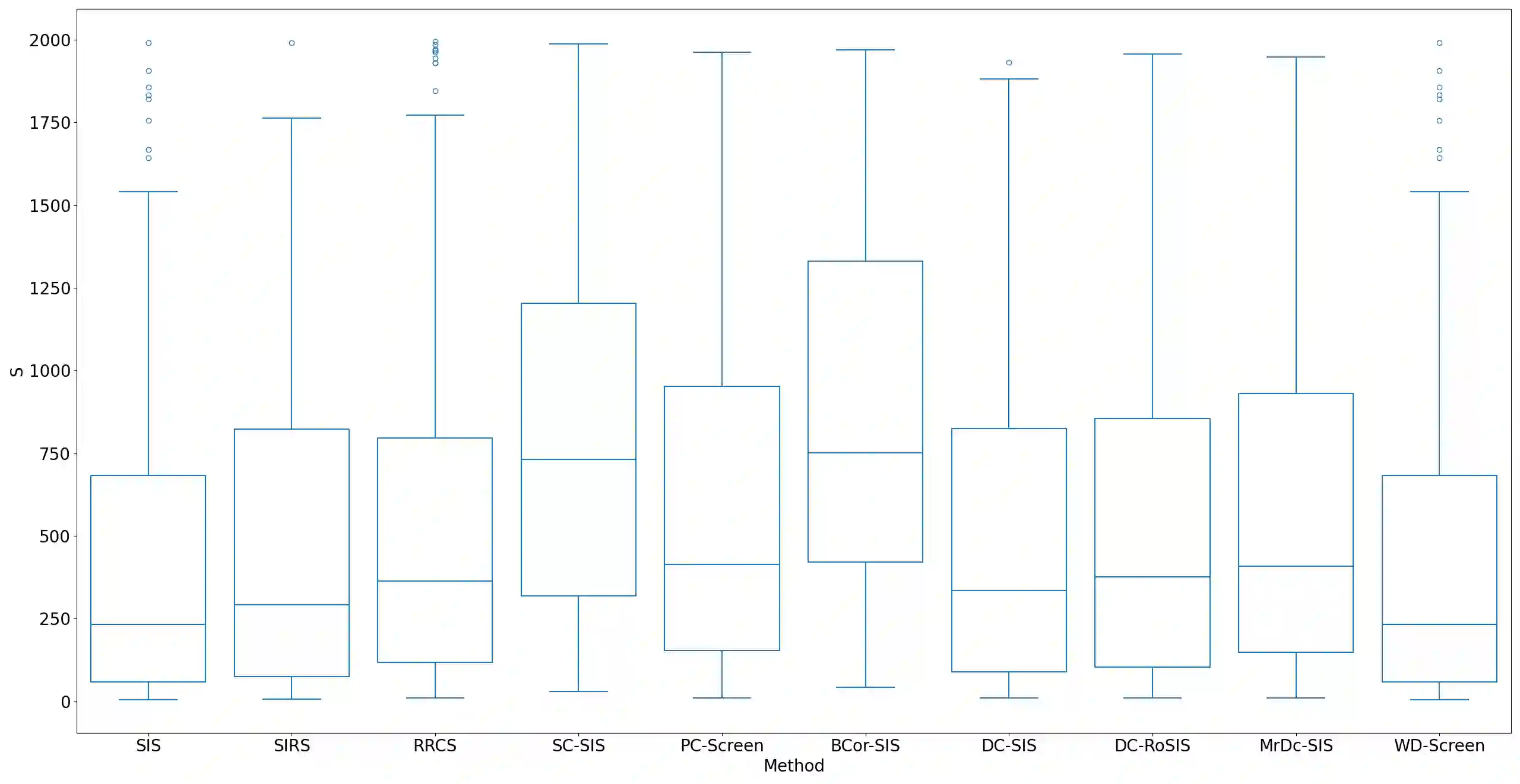
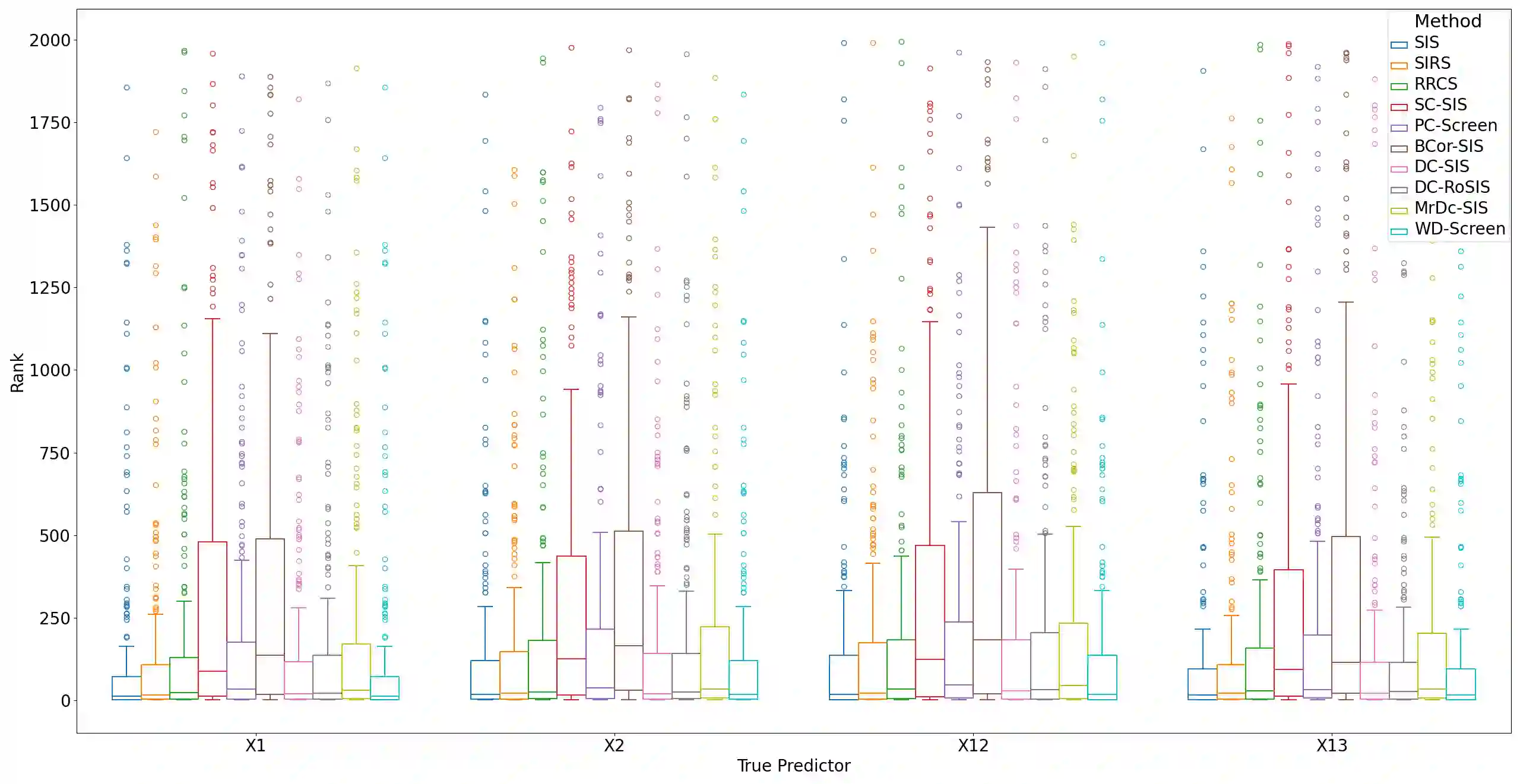
Lung adenocarcinoma (LUAD) is characterized by substantial genetic heterogeneity, posing challenges in identifying reliable biomarkers for improved diagnosis and treatment. Tumor Mutational Burden (TMB) has traditionally been regarded as a predictive biomarker, given its association with immune response and treatment efficacy. In this study, we treated TMB as a response variable to identify genes highly correlated with it, aiming to understand its genetic drivers. We conducted a thorough investigation of recent feature selection methods through extensive simulations, selecting PC-Screen, DC-SIS, and WD-Screen as top performers. These methods handle multi-omics structures effectively, and can accommodate both categorical and continuous data types at the same time for each gene. Using data from The Cancer Genome Atlas (TCGA) via cBioPortal, we combined copy number alteration (CNA), mRNA expression and DNA methylation data as multi-omics predictors and applied these methods, selecting genes consistently identified across all three methods. 13 common genes were identified, including HSD17B4, PCBD2, which show strong associations with TMB. Our multi-omics strategy and robust feature selection approach provide insights into the genetic determinants of TMB, with implications for targeted LUAD therapies.
翻译:肺腺癌(LUAD)具有显著的遗传异质性,这为识别可靠的生物标志物以改善诊断和治疗带来了挑战。肿瘤突变负荷(TMB)传统上被视为一种预测性生物标志物,因其与免疫反应和治疗效果相关。在本研究中,我们将TMB视为响应变量,以识别与其高度相关的基因,旨在理解其遗传驱动因素。我们通过大量模拟对近期特征选择方法进行了全面研究,筛选出表现最佳的PC-Screen、DC-SIS和WD-Screen方法。这些方法能有效处理多组学数据结构,并且能同时适应每个基因的分类和连续数据类型。利用通过cBioPortal获取的癌症基因组图谱(TCGA)数据,我们将拷贝数变异(CNA)、mRNA表达数据和DNA甲基化数据组合为多组学预测因子,并应用这些方法,筛选出在所有三种方法中均被一致识别的基因。共鉴定出13个共有基因,包括HSD17B4、PCBD2等,这些基因显示出与TMB的强关联性。我们的多组学策略和稳健的特征选择方法为理解TMB的遗传决定因素提供了见解,对肺腺癌的靶向治疗具有潜在意义。