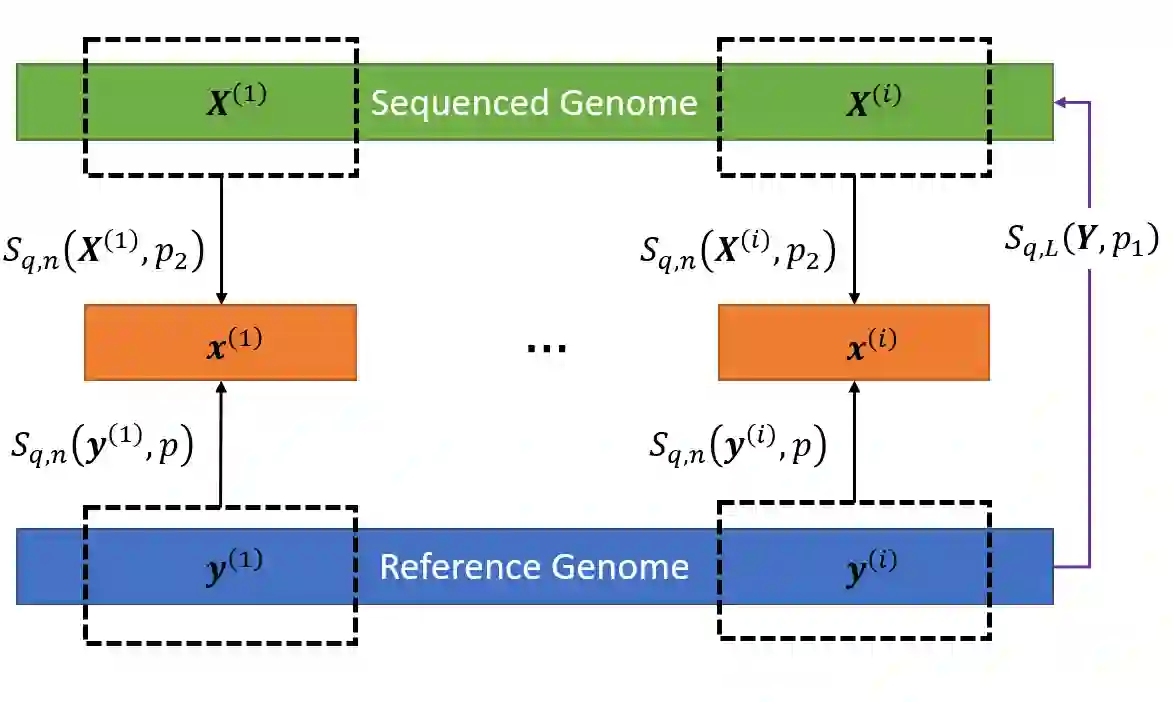
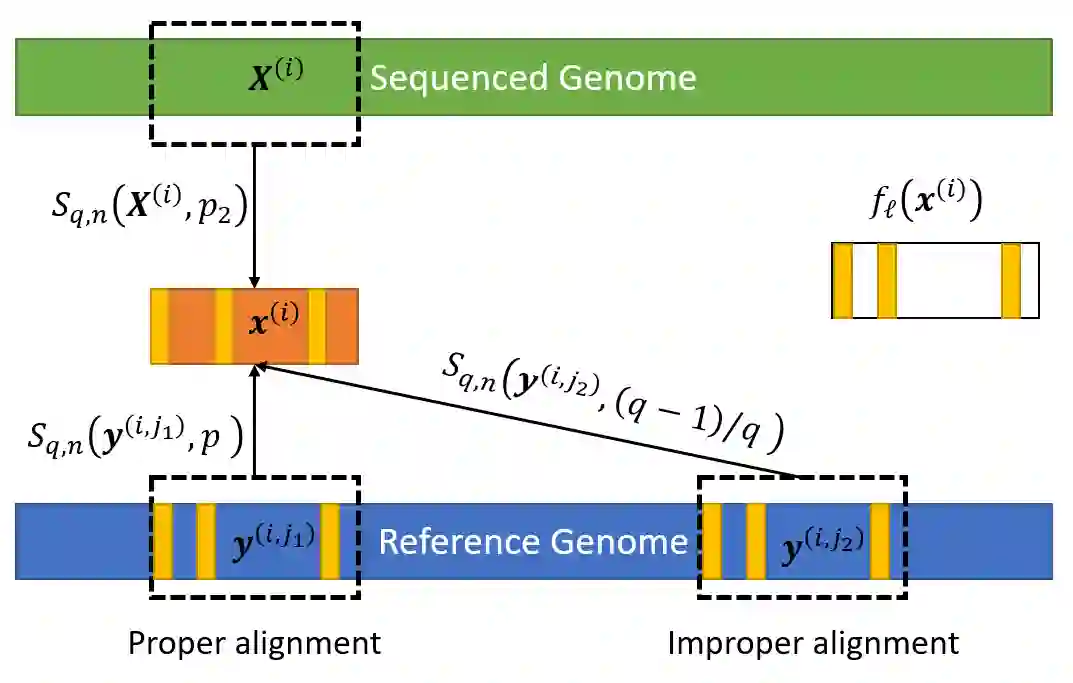
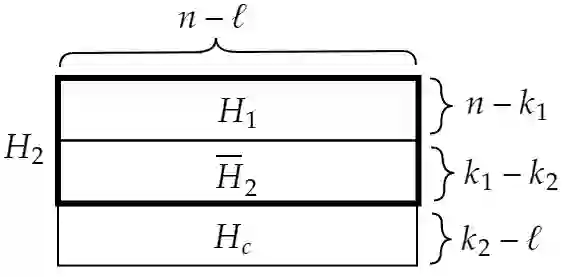
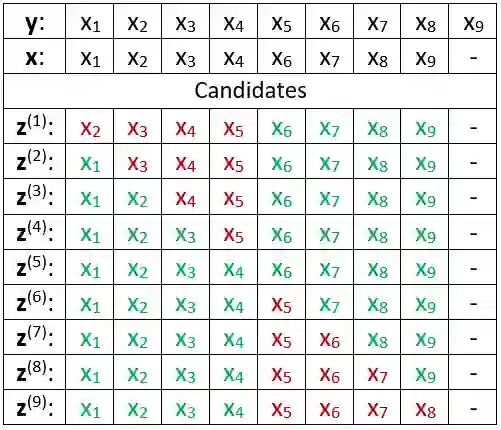
We propose a new compression scheme for genomic data given as sequence fragments called reads. The scheme uses a reference genome at the decoder side only, freeing the encoder from the burdens of storing references and performing computationally costly alignment operations. The main ingredient of the scheme is a multi-layer code construction, delivering to the decoder sufficient information to align the reads, correct their differences from the reference, validate their reconstruction, and correct reconstruction errors. The core of the method is the well-known concept of distributed source coding with decoder side information, fortified by a generalized-concatenation code construction enabling efficient embedding of all the information needed for reliable reconstruction. We first present the scheme for the case of substitution errors only between the reads and the reference, and then extend it to support reads with a single deletion and multiple substitutions. A central tool in this extension is a new distance metric that is shown analytically to improve alignment performance over existing distance metrics.
翻译:我们提出一种针对基因组序列片段(称为读取片段)的新型压缩方案。该方案仅在解码端使用参考基因组,使编码端无需承担存储参考序列及执行高计算开销比对操作的重任。方案核心为多层编码架构,能为解码端提供充足信息以完成读取比对、修正与参考序列的差异、验证重建质量并纠正重建错误。该方法的基础是经典的带解码端辅助信息的分布式信源编码理论,通过广义级联编码构造实现高效嵌入可靠重建所需的所有信息。我们首先针对读取片段与参考序列间仅存在替换错误的情形提出该方案,随后将其扩展至支持含单个删除错误与多个替换错误的读取片段。该扩展的核心工具是一种新型距离度量,理论分析表明其相较于现有距离度量能显著提升比对性能。