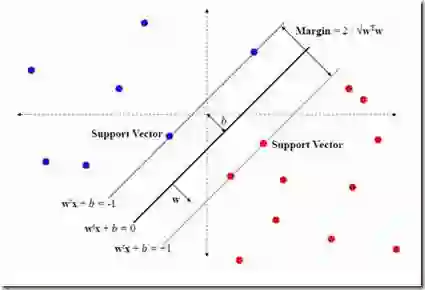
The innovation of next-generation sequencing (NGS) techniques has significantly reduced the price of genome sequencing, lowering barriers to future medical research; it is now feasible to apply genome sequencing to studies where it would have previously been cost-inefficient. Identifying damaging or pathogenic mutations in vast amounts of complex, high-dimensional genome sequencing data may be of particular interest to researchers. Thus, this paper's aims were to train machine learning models on the attributes of a genetic mutation to predict LoFtool scores (which measure a gene's intolerance to loss-of-function mutations). These attributes included, but were not limited to, the position of a mutation on a chromosome, changes in amino acids, and changes in codons caused by the mutation. Models were built using the univariate feature selection technique f-regression combined with K-nearest neighbors (KNN), Support Vector Machine (SVM), Random Sample Consensus (RANSAC), Decision Trees, Random Forest, and Extreme Gradient Boosting (XGBoost). These models were evaluated using five-fold cross-validated averages of r-squared, mean squared error, root mean squared error, mean absolute error, and explained variance. The findings of this study include the training of multiple models with testing set r-squared values of 0.97.
翻译:下一代测序(NGS)技术的创新显著降低了基因组测序的成本,为未来医学研究扫清了障碍;如今,在以往因成本效益低下而无法开展的研究中应用基因组测序已成为可能。从海量复杂、高维的基因组测序数据中识别有害或致病性突变,可能特别引起研究者的关注。因此,本文旨在基于遗传突变的特征训练机器学习模型,以预测LoFtool评分(该评分衡量基因对功能丧失突变的耐受性)。这些特征包括但不限于突变在染色体上的位置、氨基酸变化以及突变导致的密码子变化。模型采用单变量特征选择技术f-回归结合K近邻(KNN)、支持向量机(SVM)、随机采样一致性(RANSAC)、决策树、随机森林和极限梯度提升(XGBoost)构建。通过五折交叉验证的平均指标(包括R方、均方误差、均方根误差、平均绝对误差和解释方差)对模型进行评估。本研究发现包括训练了多个测试集R方值达到0.97的模型。