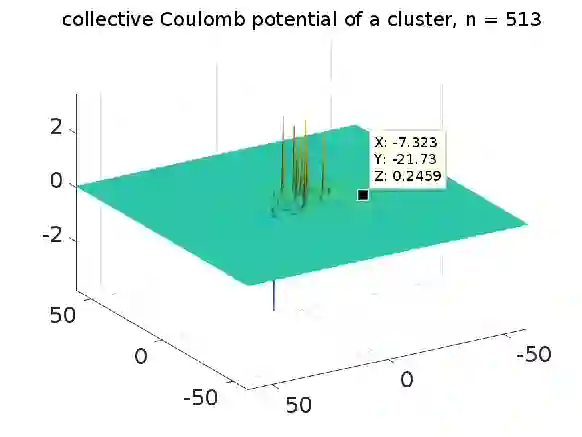
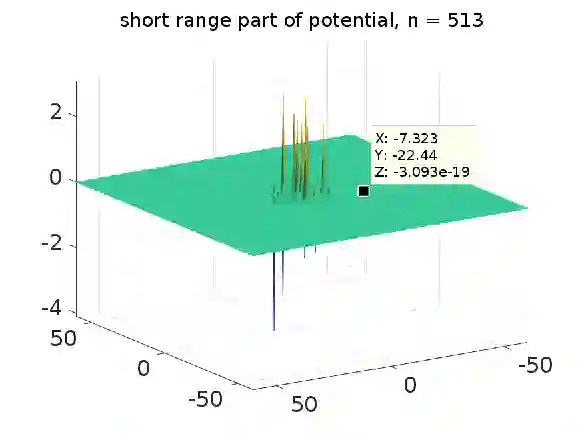
We propose and justify a new approach for fast calculation of the electrostatic interaction energy of clusters of charged particles in constrained energy minimization in the framework of rigid protein-ligand docking. Our ``blind search'' docking technique is based on the low-rank range-separated (RS) tensor-based representation of the free-space electrostatic potential of the biomolecule represented on large $n\times n\times n$ 3D grid. We show that both the collective electrostatic potential of a complex protein-ligand system and the respective electrostatic interaction energy can be calculated by tensor techniques in $O(n)$-complexity, such that the numerical cost for energy calculation only mildly (logarithmically) depends on the number of particles in the system. Moreover, tensor representation of the electrostatic potential enables usage of large 3D Cartesian grids (of the order of $n^3 \sim 10^{12}$), which could allow the accurate modeling of complexes with several large proteins. In our approach selection of the correct geometric pose predictions in the localized posing process is based on the control of van der Waals distance between the target molecular clusters. Here, we confine ourselves by constrained minimization of the energy functional by using only fast tensor-based free-space electrostatic energy recalculation for various rotations and translations of both clusters. Numerical tests of the electrostatic energy-based ``protein-ligand docking'' algorithm applied to synthetic and realistic input data present a proof of concept for rather complex particle configurations. The method may be used in the framework of the traditional stochastic or deterministic posing/docking techniques.
翻译:本文提出并论证了一种在刚性蛋白质-配体对接框架下,用于约束能量最小化中带电粒子团静电相互作用能快速计算的新方法。我们的“盲搜索”对接技术基于生物分子自由空间静电势的低秩范围分离张量表示,该表示构建于大型 $n\\times n\\times n$ 三维网格上。研究表明,复杂蛋白质-配体系统的整体静电势及相应的静电相互作用能均可通过张量技术以 $O(n)$ 复杂度计算,使得能量计算的数值成本仅微弱地(对数级)依赖于系统中的粒子数量。此外,静电势的张量表示支持使用大型三维笛卡尔网格(规模达 $n^3 \\sim 10^{12}$ 量级),从而能够精确模拟包含多个大型蛋白质的复合物。在我们的方法中,局部构象预测过程中正确几何姿态的筛选基于对目标分子团间范德华距离的控制。此处,我们通过仅使用快速张量自由空间静电能量重计算,对两个团簇的各种旋转和平移进行约束能量泛函最小化。基于静电能量的“蛋白质-配体对接”算法在合成及真实输入数据上的数值测试,为相当复杂的粒子构型提供了概念验证。该方法可应用于传统的随机或确定性构象/对接技术框架中。