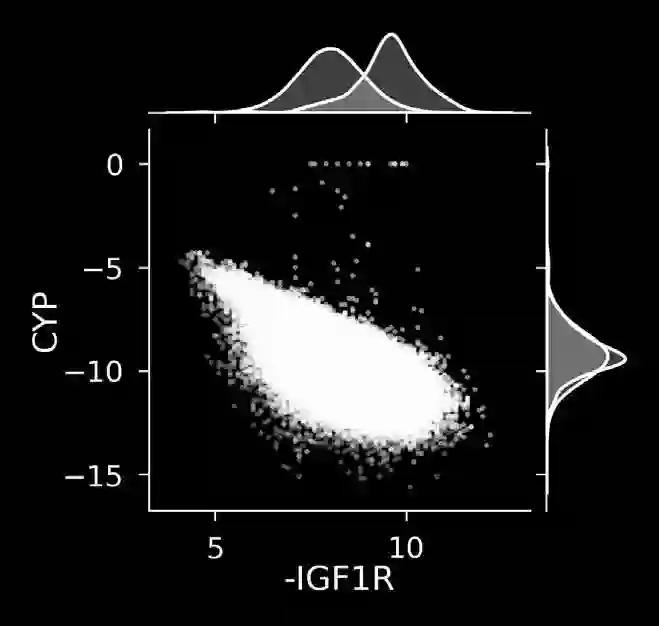
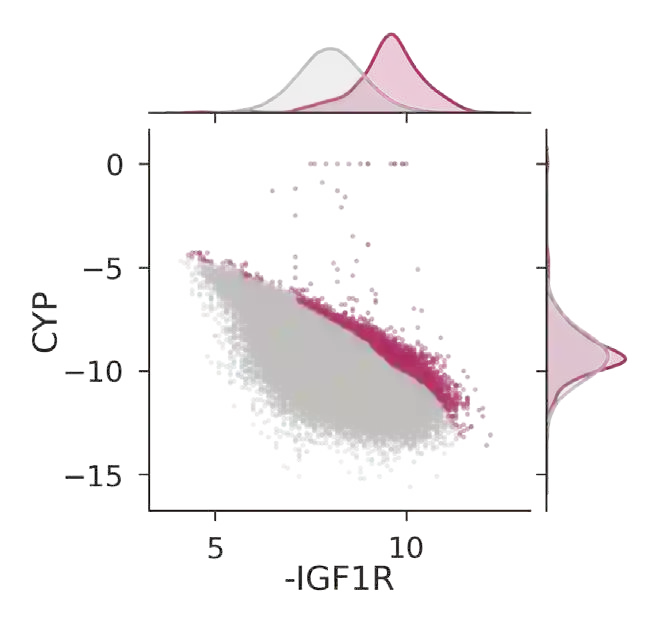
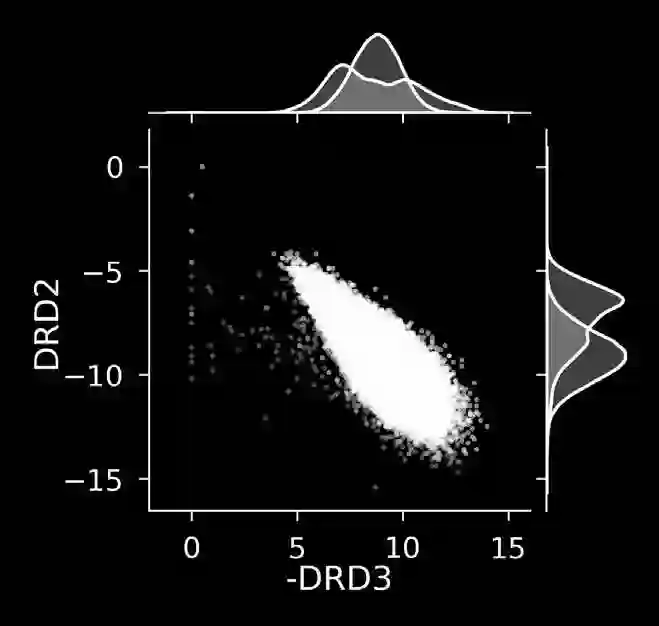
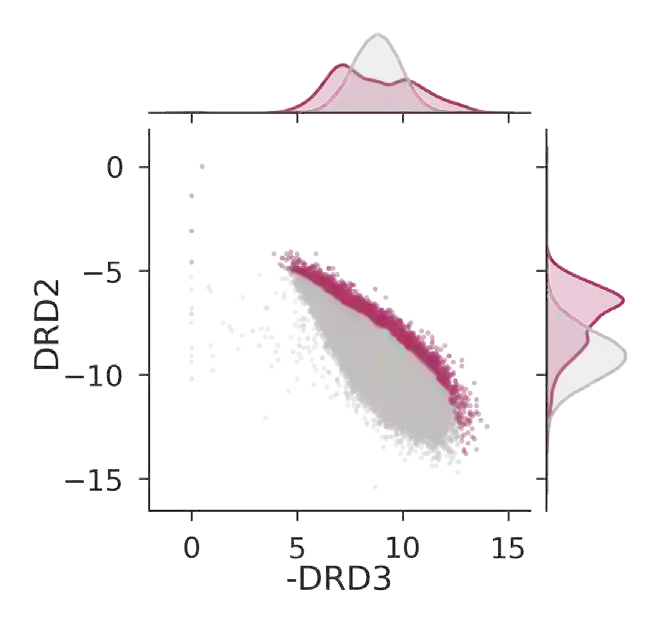
The discovery of therapeutic molecules is fundamentally a multi-objective optimization problem. One formulation of the problem is to identify molecules that simultaneously exhibit strong binding affinity for a target protein, minimal off-target interactions, and suitable pharmacokinetic properties. Inspired by prior work that uses active learning to accelerate the identification of strong binders, we implement multi-objective Bayesian optimization to reduce the computational cost of multi-property virtual screening and apply it to the identification of ligands predicted to be selective based on docking scores to on- and off-targets. We demonstrate the superiority of Pareto optimization over scalarization across three case studies. Further, we use the developed optimization tool to search a virtual library of over 4M molecules for those predicted to be selective dual inhibitors of EGFR and IGF1R, acquiring 100% of the molecules that form the library's Pareto front after exploring only 8% of the library. This workflow and associated open source software can reduce the screening burden of molecular design projects and is complementary to research aiming to improve the accuracy of binding predictions and other molecular properties.
翻译:治疗性分子的发现本质上是一个多目标优化问题。该问题的一种形式化表述是识别同时具备对靶蛋白强结合亲和力、最小脱靶相互作用以及合适药代动力学性质的分子。受先前利用主动学习加速强结合分子识别研究的启发,我们实现了多目标贝叶斯优化以减少多属性虚拟筛选的计算成本,并将其应用于基于对靶标和脱靶分子对接评分预测具有选择性的配体识别。通过三个案例研究,我们证明了帕累托优化相较于标量化的优越性。此外,我们利用所开发的优化工具在包含超过400万个分子的虚拟库中搜索预测为EGFR和IGF1R选择性双重抑制剂的分子,在仅探索库中8%的分子后,便获得了构成该库帕累托前沿的全部分子。本工作流程及相关的开源软件可减轻分子设计项目的筛选负担,并与旨在提高结合预测及其他分子性质准确性的研究形成互补。