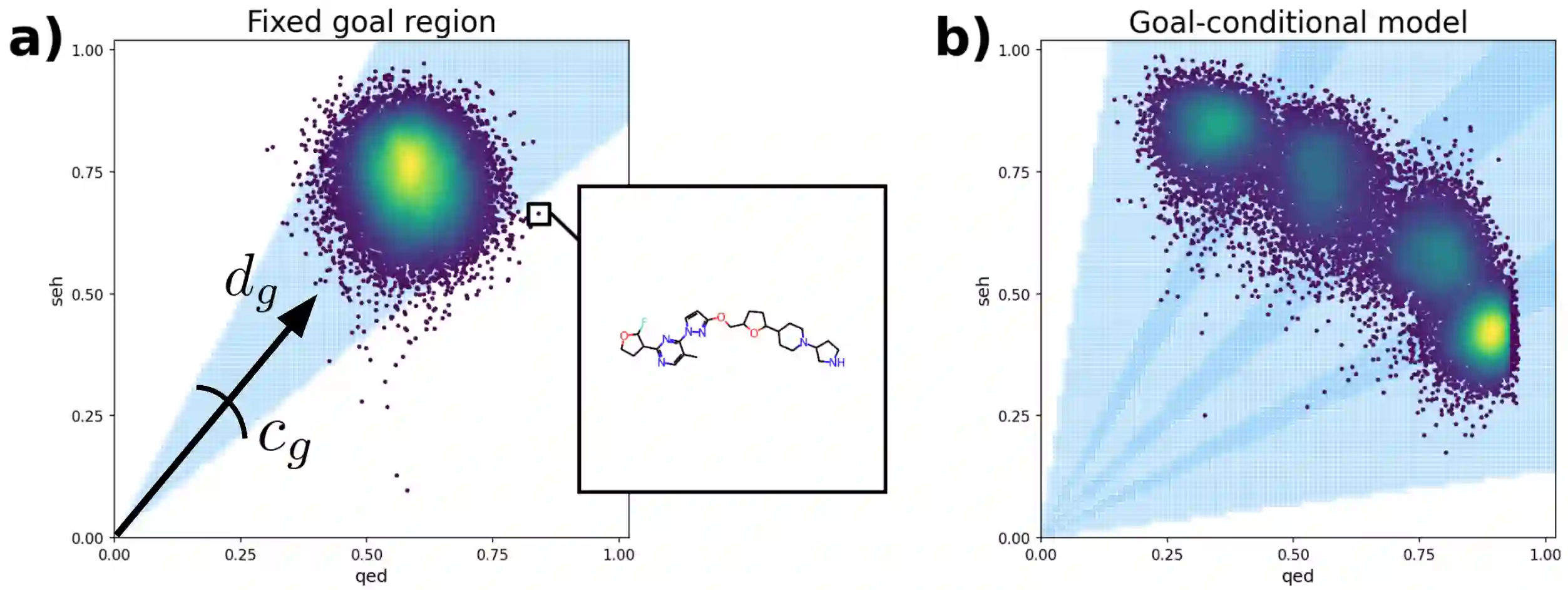
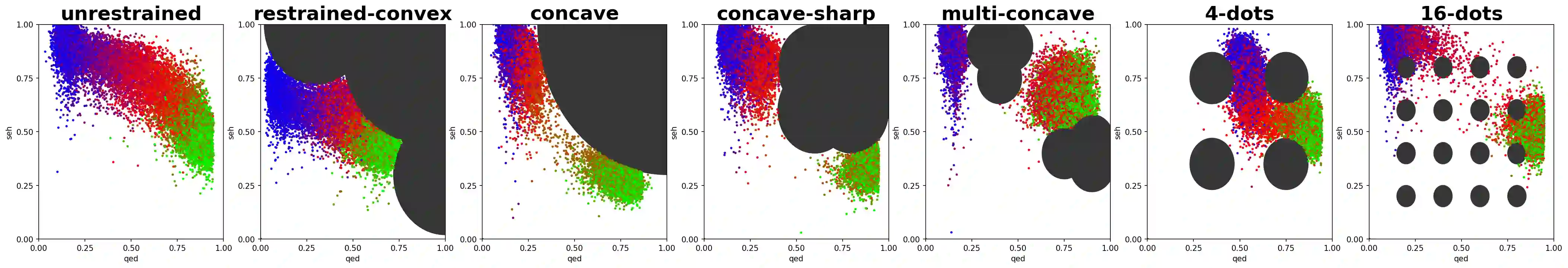
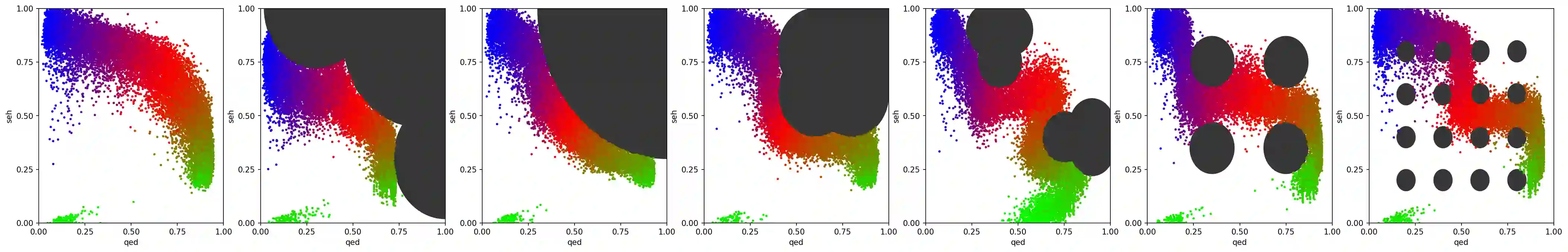
In recent years, in-silico molecular design has received much attention from the machine learning community. When designing a new compound for pharmaceutical applications, there are usually multiple properties of such molecules that need to be optimised: binding energy to the target, synthesizability, toxicity, EC50, and so on. While previous approaches have employed a scalarization scheme to turn the multi-objective problem into a preference-conditioned single objective, it has been established that this kind of reduction may produce solutions that tend to slide towards the extreme points of the objective space when presented with a problem that exhibits a concave Pareto front. In this work we experiment with an alternative formulation of goal-conditioned molecular generation to obtain a more controllable conditional model that can uniformly explore solutions along the entire Pareto front.
翻译:近年来,计算机模拟分子设计引起了机器学习领域的广泛关注。在为药物应用设计新化合物时,通常需要优化分子的多种属性:与靶点的结合能、可合成性、毒性、半最大效应浓度(EC50)等。尽管先前的方法采用标量化方案将多目标问题转化为偏好条件下的单目标问题,但已有研究表明,当面对具有凹帕累托前沿的问题时,这种简化方式产生的解往往倾向于滑向目标空间的极端点。本研究尝试采用一种替代性的目标条件分子生成框架,构建更具可控性的条件模型,使其能够沿整个帕累托前沿均匀探索解空间。