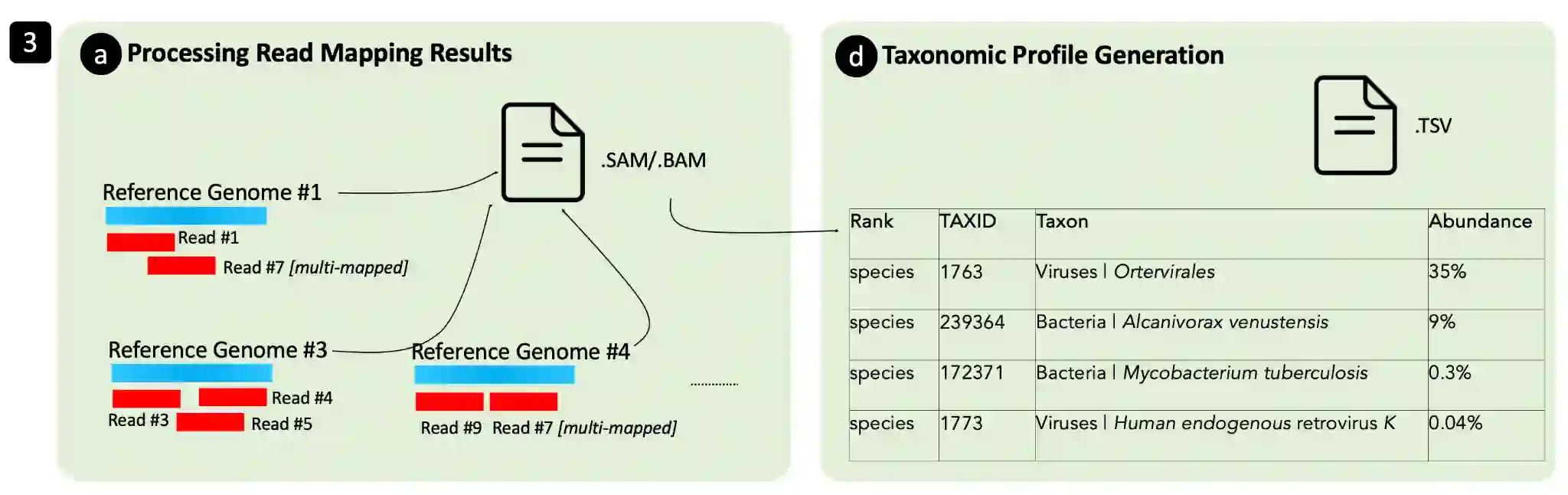
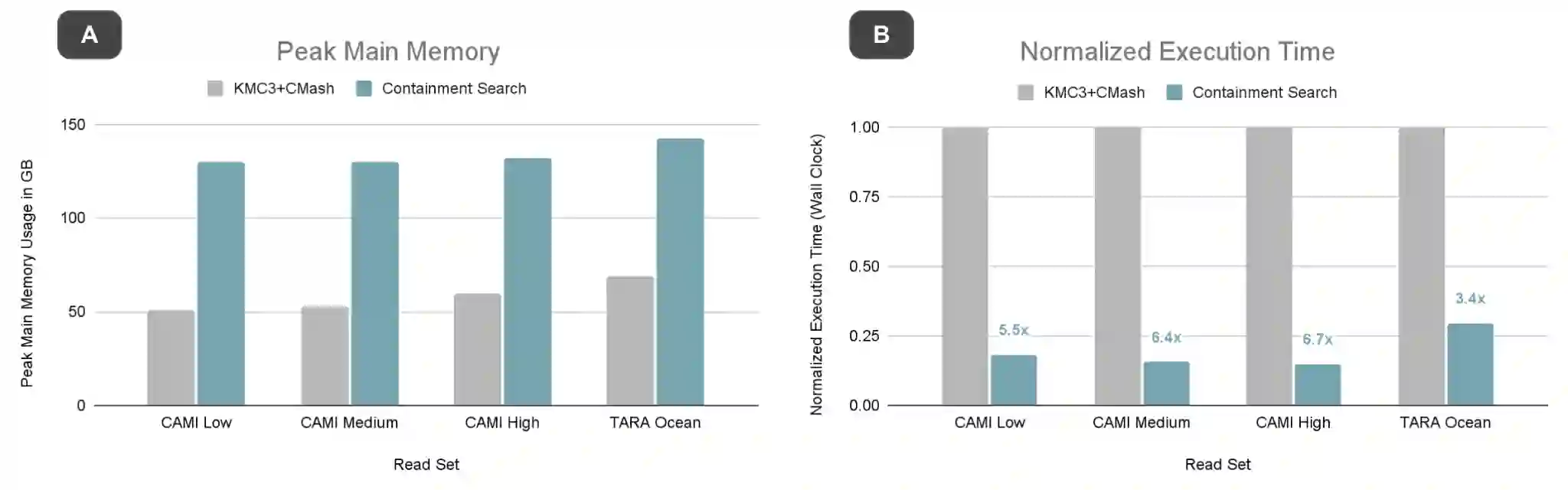
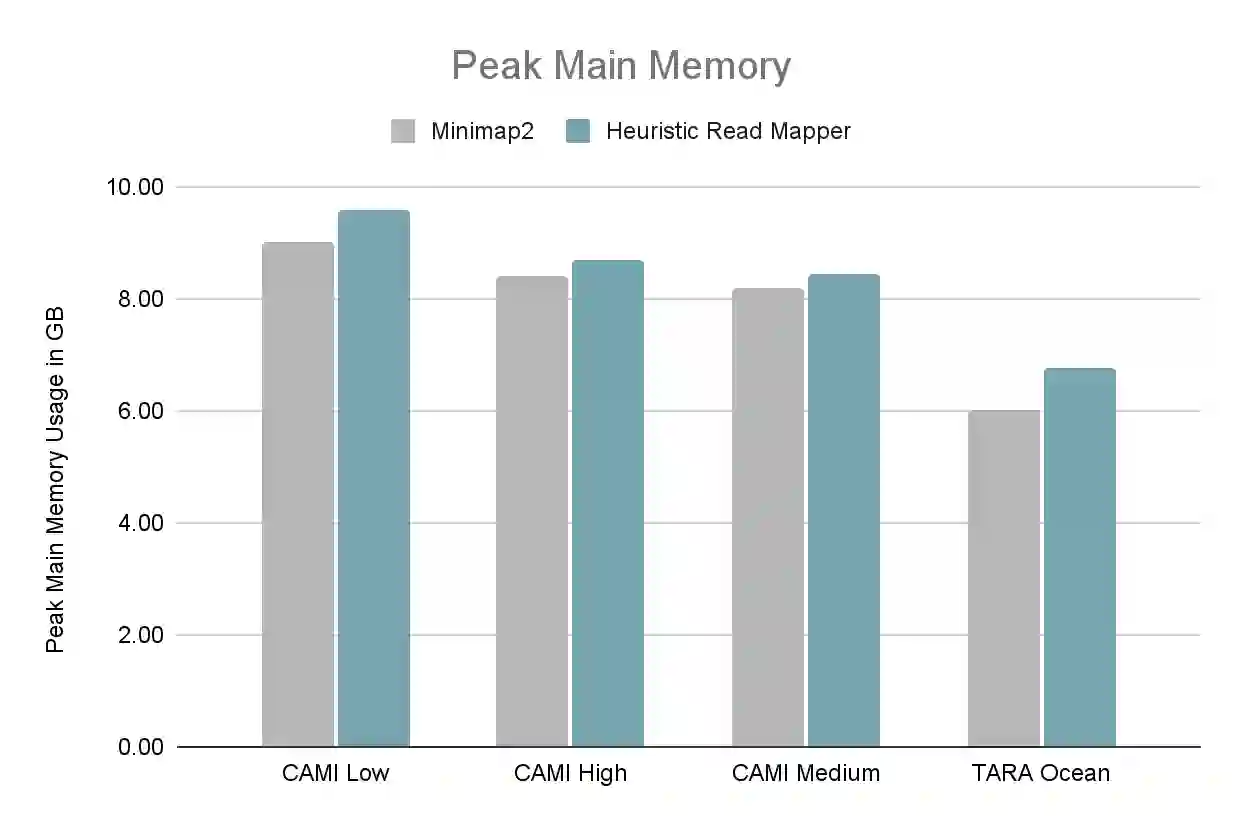
Metagenomics, the study of genome sequences of diverse organisms cohabiting in a shared environment, has experienced significant advancements across various medical and biological fields. Metagenomic analysis is crucial, for instance, in clinical applications such as infectious disease screening and the diagnosis and early detection of diseases such as cancer. A key task in metagenomics is to determine the species present in a sample and their relative abundances. Currently, the field is dominated by either alignment-based tools, which offer high accuracy but are computationally expensive, or alignment-free tools, which are fast but lack the needed accuracy for many applications. In response to this dichotomy, we introduce MetaFast, a tool based on heuristics, to achieve a fundamental improvement in accuracy-runtime tradeoff over existing methods. MetaFast delivers accuracy comparable to the alignment-based and highly accurate tool Metalign but with significantly enhanced efficiency. In MetaFast, we accelerate memory-frugal reference database indexing and filtering. We further employ heuristics to accelerate read mapping. Our evaluation demonstrates that MetaFast achieves a 4x speedup over Metalign without compromising accuracy. MetaFast is publicly available on: https://github.com/CMU-SAFARI/MetaFast.
翻译:宏基因组学是研究共享环境中多种共存生物基因组序列的学科,已在医学和生物学多个领域取得显著进展。例如,在传染病筛查及癌症等疾病的诊断与早期检测等临床应用场景中,宏基因组分析至关重要。宏基因组学的核心任务之一是确定样本中存在的物种及其相对丰度。当前,该领域主要采用两类工具:基于比对的方法虽高精度但计算成本高昂,而免比对方法虽快速但缺乏许多应用所需的精度。针对这一两难问题,我们提出了基于启发式策略的MetaFast工具,旨在从根本上改善现有方法在精度与运行时间之间的权衡。MetaFast在保持与高精度比对工具Metalign相当的精度的同时,显著提升了效率。在MetaFast中,我们加速了内存节约型参考数据库的索引构建与过滤,并进一步采用启发式算法加速读取映射。评估表明,MetaFast在不牺牲精度的情况下实现了比Metalign快4倍的加速。MetaFast已公开于:https://github.com/CMU-SAFARI/MetaFast。